Tania Isabella Aravena, Elizabeth Valdés, Nicolás Ayala, Vívian D'Afonseca
{"title":"预测甲基转移酶组蛋白基因遗传改变在肝癌中的作用的计算方法。","authors":"Tania Isabella Aravena, Elizabeth Valdés, Nicolás Ayala, Vívian D'Afonseca","doi":"10.1177/11769351231161480","DOIUrl":null,"url":null,"abstract":"<p><p>Histone methyltransferases (HMTs) comprise a subclass of epigenetic regulators. Dysregulation of these enzymes results in aberrant epigenetic regulation, commonly observed in various tumor types, including hepatocellular adenocarcinoma (HCC). Probably, these epigenetic changes could lead to tumorigenesis processes. To predict how histone methyltransferase genes and their genetic alterations (somatic mutations, somatic copy number alterations, and gene expression changes) are involved in hepatocellular adenocarcinoma processes, we performed an integrated computational analysis of genetic alterations in 50 HMT genes present in hepatocellular adenocarcinoma. Biological data were obtained through the public repository with 360 samples from patients with hepatocellular carcinoma. Through these biological data, we identified 10 HMT genes (<i>SETDB1, ASH1L, SMYD2, SMYD3, EHMT2, SETD3, PRDM14, PRDM16, KMT2C</i>, and <i>NSD3</i>) with a significant genetic alteration rate (14%) within 360 samples. Of these 10 HMT genes, <i>KMT2C</i> and <i>ASH1L</i> have the highest mutation rate in HCC samples, 5.6% and 2.8%, respectively. Regarding somatic copy number alteration, <i>ASH1L</i> and <i>SETDB1</i> are amplified in several samples, while <i>SETD3, PRDM14</i>, and <i>NSD3</i> showed a high rate of large deletion. Finally, <i>SETDB1, SETD3, PRDM14</i>, and <i>NSD3</i> could play an important role in the progression of hepatocellular adenocarcinoma since alterations in these genes lead to a decrease in patient survival, unlike patients who present these genes without genetic alterations. Our computational analysis provides new insights that help to understand how HMTs are associated with hepatocellular carcinoma, as well as provide a basis for future experimental investigations using HMTs as genetic targets against hepatocellular carcinoma.</p>","PeriodicalId":35418,"journal":{"name":"Cancer Informatics","volume":"22 ","pages":"11769351231161480"},"PeriodicalIF":2.4000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/b4/10.1177_11769351231161480.PMC10064455.pdf","citationCount":"1","resultStr":"{\"title\":\"A Computational Approach to Predict the Role of Genetic Alterations in Methyltransferase Histones Genes With Implications in Liver Cancer.\",\"authors\":\"Tania Isabella Aravena, Elizabeth Valdés, Nicolás Ayala, Vívian D'Afonseca\",\"doi\":\"10.1177/11769351231161480\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Histone methyltransferases (HMTs) comprise a subclass of epigenetic regulators. Dysregulation of these enzymes results in aberrant epigenetic regulation, commonly observed in various tumor types, including hepatocellular adenocarcinoma (HCC). Probably, these epigenetic changes could lead to tumorigenesis processes. To predict how histone methyltransferase genes and their genetic alterations (somatic mutations, somatic copy number alterations, and gene expression changes) are involved in hepatocellular adenocarcinoma processes, we performed an integrated computational analysis of genetic alterations in 50 HMT genes present in hepatocellular adenocarcinoma. Biological data were obtained through the public repository with 360 samples from patients with hepatocellular carcinoma. Through these biological data, we identified 10 HMT genes (<i>SETDB1, ASH1L, SMYD2, SMYD3, EHMT2, SETD3, PRDM14, PRDM16, KMT2C</i>, and <i>NSD3</i>) with a significant genetic alteration rate (14%) within 360 samples. Of these 10 HMT genes, <i>KMT2C</i> and <i>ASH1L</i> have the highest mutation rate in HCC samples, 5.6% and 2.8%, respectively. Regarding somatic copy number alteration, <i>ASH1L</i> and <i>SETDB1</i> are amplified in several samples, while <i>SETD3, PRDM14</i>, and <i>NSD3</i> showed a high rate of large deletion. Finally, <i>SETDB1, SETD3, PRDM14</i>, and <i>NSD3</i> could play an important role in the progression of hepatocellular adenocarcinoma since alterations in these genes lead to a decrease in patient survival, unlike patients who present these genes without genetic alterations. Our computational analysis provides new insights that help to understand how HMTs are associated with hepatocellular carcinoma, as well as provide a basis for future experimental investigations using HMTs as genetic targets against hepatocellular carcinoma.</p>\",\"PeriodicalId\":35418,\"journal\":{\"name\":\"Cancer Informatics\",\"volume\":\"22 \",\"pages\":\"11769351231161480\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1e/b4/10.1177_11769351231161480.PMC10064455.pdf\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cancer Informatics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11769351231161480\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cancer Informatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11769351231161480","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
A Computational Approach to Predict the Role of Genetic Alterations in Methyltransferase Histones Genes With Implications in Liver Cancer.
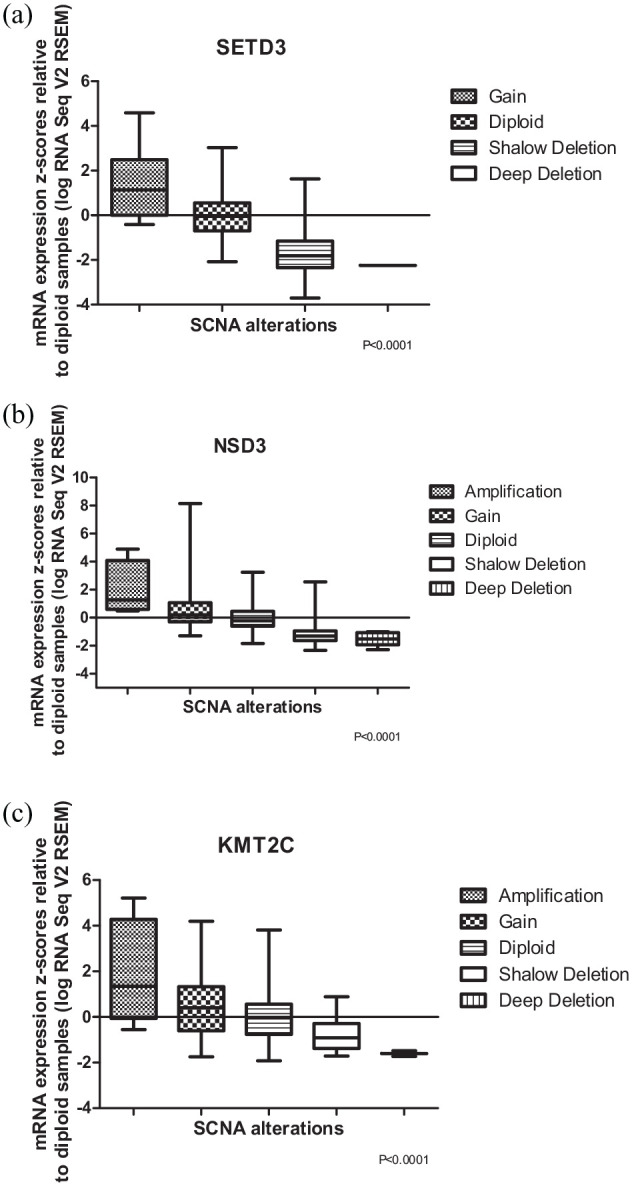
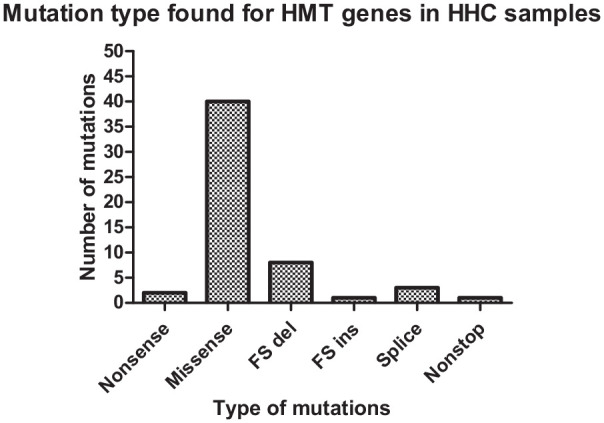
Histone methyltransferases (HMTs) comprise a subclass of epigenetic regulators. Dysregulation of these enzymes results in aberrant epigenetic regulation, commonly observed in various tumor types, including hepatocellular adenocarcinoma (HCC). Probably, these epigenetic changes could lead to tumorigenesis processes. To predict how histone methyltransferase genes and their genetic alterations (somatic mutations, somatic copy number alterations, and gene expression changes) are involved in hepatocellular adenocarcinoma processes, we performed an integrated computational analysis of genetic alterations in 50 HMT genes present in hepatocellular adenocarcinoma. Biological data were obtained through the public repository with 360 samples from patients with hepatocellular carcinoma. Through these biological data, we identified 10 HMT genes (SETDB1, ASH1L, SMYD2, SMYD3, EHMT2, SETD3, PRDM14, PRDM16, KMT2C, and NSD3) with a significant genetic alteration rate (14%) within 360 samples. Of these 10 HMT genes, KMT2C and ASH1L have the highest mutation rate in HCC samples, 5.6% and 2.8%, respectively. Regarding somatic copy number alteration, ASH1L and SETDB1 are amplified in several samples, while SETD3, PRDM14, and NSD3 showed a high rate of large deletion. Finally, SETDB1, SETD3, PRDM14, and NSD3 could play an important role in the progression of hepatocellular adenocarcinoma since alterations in these genes lead to a decrease in patient survival, unlike patients who present these genes without genetic alterations. Our computational analysis provides new insights that help to understand how HMTs are associated with hepatocellular carcinoma, as well as provide a basis for future experimental investigations using HMTs as genetic targets against hepatocellular carcinoma.
期刊介绍:
The field of cancer research relies on advances in many other disciplines, including omics technology, mass spectrometry, radio imaging, computer science, and biostatistics. Cancer Informatics provides open access to peer-reviewed high-quality manuscripts reporting bioinformatics analysis of molecular genetics and/or clinical data pertaining to cancer, emphasizing the use of machine learning, artificial intelligence, statistical algorithms, advanced imaging techniques, data visualization, and high-throughput technologies. As the leading journal dedicated exclusively to the report of the use of computational methods in cancer research and practice, Cancer Informatics leverages methodological improvements in systems biology, genomics, proteomics, metabolomics, and molecular biochemistry into the fields of cancer detection, treatment, classification, risk-prediction, prevention, outcome, and modeling.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
