Mohammad Lotfollahi, Anna Klimovskaia Susmelj, Carlo De Donno, Leon Hetzel, Yuge Ji, Ignacio L Ibarra, Sanjay R Srivatsan, Mohsen Naghipourfar, Riza M Daza, Beth Martin, Jay Shendure, Jose L McFaline-Figueroa, Pierre Boyeau, F Alexander Wolf, Nafissa Yakubova, Stephan Günnemann, Cole Trapnell, David Lopez-Paz, Fabian J Theis
{"title":"在高通量筛选中预测细胞对复杂扰动的反应。","authors":"Mohammad Lotfollahi, Anna Klimovskaia Susmelj, Carlo De Donno, Leon Hetzel, Yuge Ji, Ignacio L Ibarra, Sanjay R Srivatsan, Mohsen Naghipourfar, Riza M Daza, Beth Martin, Jay Shendure, Jose L McFaline-Figueroa, Pierre Boyeau, F Alexander Wolf, Nafissa Yakubova, Stephan Günnemann, Cole Trapnell, David Lopez-Paz, Fabian J Theis","doi":"10.15252/msb.202211517","DOIUrl":null,"url":null,"abstract":"<p><p>Recent advances in multiplexed single-cell transcriptomics experiments facilitate the high-throughput study of drug and genetic perturbations. However, an exhaustive exploration of the combinatorial perturbation space is experimentally unfeasible. Therefore, computational methods are needed to predict, interpret, and prioritize perturbations. Here, we present the compositional perturbation autoencoder (CPA), which combines the interpretability of linear models with the flexibility of deep-learning approaches for single-cell response modeling. CPA learns to in silico predict transcriptional perturbation response at the single-cell level for unseen dosages, cell types, time points, and species. Using newly generated single-cell drug combination data, we validate that CPA can predict unseen drug combinations while outperforming baseline models. Additionally, the architecture's modularity enables incorporating the chemical representation of the drugs, allowing the prediction of cellular response to completely unseen drugs. Furthermore, CPA is also applicable to genetic combinatorial screens. We demonstrate this by imputing in silico 5,329 missing combinations (97.6% of all possibilities) in a single-cell Perturb-seq experiment with diverse genetic interactions. We envision CPA will facilitate efficient experimental design and hypothesis generation by enabling in silico response prediction at the single-cell level and thus accelerate therapeutic applications using single-cell technologies.</p>","PeriodicalId":18906,"journal":{"name":"Molecular Systems Biology","volume":"19 6","pages":"e11517"},"PeriodicalIF":7.7000,"publicationDate":"2023-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10258562/pdf/","citationCount":"16","resultStr":"{\"title\":\"Predicting cellular responses to complex perturbations in high-throughput screens.\",\"authors\":\"Mohammad Lotfollahi, Anna Klimovskaia Susmelj, Carlo De Donno, Leon Hetzel, Yuge Ji, Ignacio L Ibarra, Sanjay R Srivatsan, Mohsen Naghipourfar, Riza M Daza, Beth Martin, Jay Shendure, Jose L McFaline-Figueroa, Pierre Boyeau, F Alexander Wolf, Nafissa Yakubova, Stephan Günnemann, Cole Trapnell, David Lopez-Paz, Fabian J Theis\",\"doi\":\"10.15252/msb.202211517\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Recent advances in multiplexed single-cell transcriptomics experiments facilitate the high-throughput study of drug and genetic perturbations. However, an exhaustive exploration of the combinatorial perturbation space is experimentally unfeasible. Therefore, computational methods are needed to predict, interpret, and prioritize perturbations. Here, we present the compositional perturbation autoencoder (CPA), which combines the interpretability of linear models with the flexibility of deep-learning approaches for single-cell response modeling. CPA learns to in silico predict transcriptional perturbation response at the single-cell level for unseen dosages, cell types, time points, and species. Using newly generated single-cell drug combination data, we validate that CPA can predict unseen drug combinations while outperforming baseline models. Additionally, the architecture's modularity enables incorporating the chemical representation of the drugs, allowing the prediction of cellular response to completely unseen drugs. Furthermore, CPA is also applicable to genetic combinatorial screens. We demonstrate this by imputing in silico 5,329 missing combinations (97.6% of all possibilities) in a single-cell Perturb-seq experiment with diverse genetic interactions. We envision CPA will facilitate efficient experimental design and hypothesis generation by enabling in silico response prediction at the single-cell level and thus accelerate therapeutic applications using single-cell technologies.</p>\",\"PeriodicalId\":18906,\"journal\":{\"name\":\"Molecular Systems Biology\",\"volume\":\"19 6\",\"pages\":\"e11517\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2023-06-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10258562/pdf/\",\"citationCount\":\"16\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.15252/msb.202211517\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.15252/msb.202211517","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Predicting cellular responses to complex perturbations in high-throughput screens.
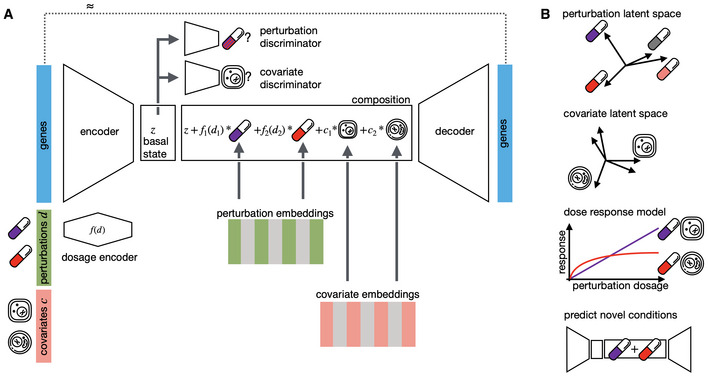
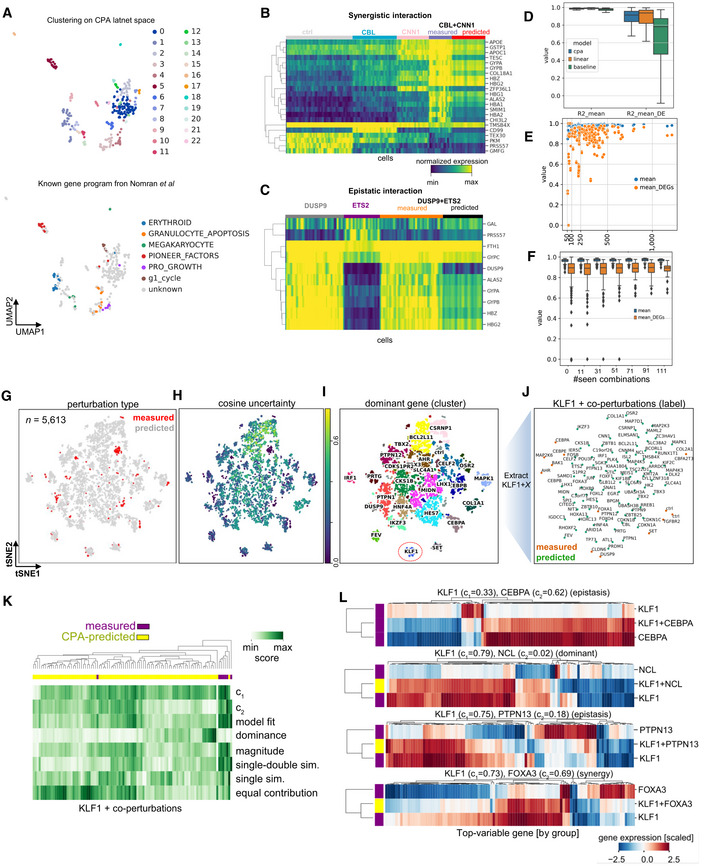
Recent advances in multiplexed single-cell transcriptomics experiments facilitate the high-throughput study of drug and genetic perturbations. However, an exhaustive exploration of the combinatorial perturbation space is experimentally unfeasible. Therefore, computational methods are needed to predict, interpret, and prioritize perturbations. Here, we present the compositional perturbation autoencoder (CPA), which combines the interpretability of linear models with the flexibility of deep-learning approaches for single-cell response modeling. CPA learns to in silico predict transcriptional perturbation response at the single-cell level for unseen dosages, cell types, time points, and species. Using newly generated single-cell drug combination data, we validate that CPA can predict unseen drug combinations while outperforming baseline models. Additionally, the architecture's modularity enables incorporating the chemical representation of the drugs, allowing the prediction of cellular response to completely unseen drugs. Furthermore, CPA is also applicable to genetic combinatorial screens. We demonstrate this by imputing in silico 5,329 missing combinations (97.6% of all possibilities) in a single-cell Perturb-seq experiment with diverse genetic interactions. We envision CPA will facilitate efficient experimental design and hypothesis generation by enabling in silico response prediction at the single-cell level and thus accelerate therapeutic applications using single-cell technologies.
期刊介绍:
Systems biology is a field that aims to understand complex biological systems by studying their components and how they interact. It is an integrative discipline that seeks to explain the properties and behavior of these systems.
Molecular Systems Biology is a scholarly journal that publishes top-notch research in the areas of systems biology, synthetic biology, and systems medicine. It is an open access journal, meaning that its content is freely available to readers, and it is peer-reviewed to ensure the quality of the published work.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
