Rachel Z. Blumhagen, David A. Schwartz, Carl D. Langefeld, Tasha E. Fingerlin
{"title":"利用随机森林重要性测度识别聚集检验中有影响的罕见变异","authors":"Rachel Z. Blumhagen, David A. Schwartz, Carl D. Langefeld, Tasha E. Fingerlin","doi":"10.1111/ahg.12509","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n \n <p>Aggregate tests of rare variants are often employed to identify associated regions compared to sequentially testing each individual variant. When an aggregate test is significant, it is of interest to identify which rare variants are “driving” the association. We recently developed the rare variant influential filtering tool (RIFT) to identify influential rare variants and showed RIFT had higher true positive rates compared to other published methods. Here we use importance measures from the standard random forest (RF) and variable importance weighted RF (vi-RF) to identify influential variants. For very rare variants (minor allele frequency [MAF] < 0.001), the vi-RF:Accuracy method had the highest median true positive rate (TPR = 0.24; interquartile range [IQR]: 0.13, 0.42) followed by the RF:Accuracy method (TPR = 0.16; IQR: 0.07, 0.33) and both were superior to RIFT (TPR = 0.05; IQR: 0.02, 0.15). Among uncommon variants (0.001 < MAF < 0.03), the RF methods had higher true positive rates than RIFT while observing comparable false positive rates. Finally, we applied the RF methods to a targeted resequencing study in idiopathic pulmonary fibrosis (IPF), in which the vi-RF approach identified eight and seven variants in <i>TERT</i> and <i>FAM13A</i>, respectively. In summary, the vi-RF provides an improved, objective approach to identifying influential variants following a significant aggregate test. We have expanded our previously developed R package RIFT to include the random forest methods.</p>\n </section>\n </div>","PeriodicalId":8085,"journal":{"name":"Annals of Human Genetics","volume":"87 4","pages":"184-195"},"PeriodicalIF":1.2000,"publicationDate":"2023-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ahg.12509","citationCount":"0","resultStr":"{\"title\":\"Identification of influential rare variants in aggregate testing using random forest importance measures\",\"authors\":\"Rachel Z. Blumhagen, David A. Schwartz, Carl D. Langefeld, Tasha E. Fingerlin\",\"doi\":\"10.1111/ahg.12509\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <section>\\n \\n \\n <p>Aggregate tests of rare variants are often employed to identify associated regions compared to sequentially testing each individual variant. When an aggregate test is significant, it is of interest to identify which rare variants are “driving” the association. We recently developed the rare variant influential filtering tool (RIFT) to identify influential rare variants and showed RIFT had higher true positive rates compared to other published methods. Here we use importance measures from the standard random forest (RF) and variable importance weighted RF (vi-RF) to identify influential variants. For very rare variants (minor allele frequency [MAF] < 0.001), the vi-RF:Accuracy method had the highest median true positive rate (TPR = 0.24; interquartile range [IQR]: 0.13, 0.42) followed by the RF:Accuracy method (TPR = 0.16; IQR: 0.07, 0.33) and both were superior to RIFT (TPR = 0.05; IQR: 0.02, 0.15). Among uncommon variants (0.001 < MAF < 0.03), the RF methods had higher true positive rates than RIFT while observing comparable false positive rates. Finally, we applied the RF methods to a targeted resequencing study in idiopathic pulmonary fibrosis (IPF), in which the vi-RF approach identified eight and seven variants in <i>TERT</i> and <i>FAM13A</i>, respectively. In summary, the vi-RF provides an improved, objective approach to identifying influential variants following a significant aggregate test. We have expanded our previously developed R package RIFT to include the random forest methods.</p>\\n </section>\\n </div>\",\"PeriodicalId\":8085,\"journal\":{\"name\":\"Annals of Human Genetics\",\"volume\":\"87 4\",\"pages\":\"184-195\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2023-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/ahg.12509\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Annals of Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/ahg.12509\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/ahg.12509","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Identification of influential rare variants in aggregate testing using random forest importance measures
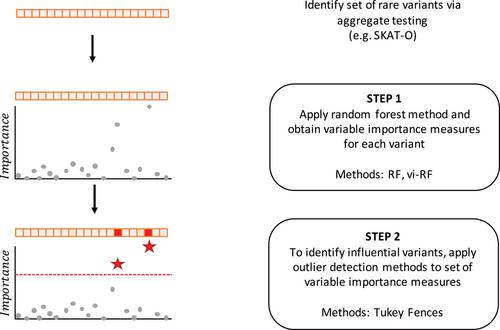
Aggregate tests of rare variants are often employed to identify associated regions compared to sequentially testing each individual variant. When an aggregate test is significant, it is of interest to identify which rare variants are “driving” the association. We recently developed the rare variant influential filtering tool (RIFT) to identify influential rare variants and showed RIFT had higher true positive rates compared to other published methods. Here we use importance measures from the standard random forest (RF) and variable importance weighted RF (vi-RF) to identify influential variants. For very rare variants (minor allele frequency [MAF] < 0.001), the vi-RF:Accuracy method had the highest median true positive rate (TPR = 0.24; interquartile range [IQR]: 0.13, 0.42) followed by the RF:Accuracy method (TPR = 0.16; IQR: 0.07, 0.33) and both were superior to RIFT (TPR = 0.05; IQR: 0.02, 0.15). Among uncommon variants (0.001 < MAF < 0.03), the RF methods had higher true positive rates than RIFT while observing comparable false positive rates. Finally, we applied the RF methods to a targeted resequencing study in idiopathic pulmonary fibrosis (IPF), in which the vi-RF approach identified eight and seven variants in TERT and FAM13A, respectively. In summary, the vi-RF provides an improved, objective approach to identifying influential variants following a significant aggregate test. We have expanded our previously developed R package RIFT to include the random forest methods.
期刊介绍:
Annals of Human Genetics publishes material directly concerned with human genetics or the application of scientific principles and techniques to any aspect of human inheritance. Papers that describe work on other species that may be relevant to human genetics will also be considered. Mathematical models should include examples of application to data where possible.
Authors are welcome to submit Supporting Information, such as data sets or additional figures or tables, that will not be published in the print edition of the journal, but which will be viewable via the online edition and stored on the website.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
