{"title":"转化浆母细胞淋巴瘤表现为明显的淋巴细胞增多和自发肿瘤溶解综合征。","authors":"Yannis Hadjiyannis, Cecelia Miller, Norris I Hollie, Jayalakshmi Balakrishna, Francesca Cottini","doi":"10.14740/jh1067","DOIUrl":null,"url":null,"abstract":"<p><p>The clinicopathology entity of plasmablastic lymphoma (PBL), despite broad recognition by the World Health Organization (WHO), represents a diagnostic challenge due to its overlapping features and scarce occurrence. Often, PBL arises in immunodeficient, elderly male patients, most notably those who are human immunodeficiency virus (HIV)-positive. More infrequent, cases of transformed PBL (tPBL) evolved from another hematologic disease have been identified. Herein, we describe a case of a 65-year-old male transferred from a neighboring hospital with pronounced lymphocytosis and spontaneous tumor lysis syndrome (sTLS) presumed to be chronic lymphocytic leukemia (CLL). Utilizing a complete clinical, morphologic, immunophenotypic, and molecular evaluation, we arrived at a final diagnosis of tPBL with sTLS, suspected to have evolved from the <i>NF-κB/NOTCH/KLF2</i> (NNK) genetic cluster of splenic marginal zone lymphoma (SMZL) (NNK-SMZL), a potential transformation and presentation, to our knowledge, not previously reported. However, definitive clonality testing was not performed. In this report, we also outline the diagnostic and educational considerations we faced in discerning tPBL from other more common B-cell malignancies which can present similarly, such as CLL, mantle cell lymphoma, or plasmablastic myeloma. We summarize recently reported molecular, prognostic, and therapeutic considerations for the treatment and recognition of PBL, including the successful implementation, in our patient, of bortezomib to an EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) regimen with prophylactic intrathecal methotrexate, who has since achieved complete remission (CR) and entered clinical surveillance. Lastly, this report briefly highlights the challenge we faced in this area of hematologic typification that necessitates additional review and discussion by the WHO: tPBL with potential double-hit cytogenetic versus double-hit lymphoma with a plasmablastic phenotype.</p>","PeriodicalId":15964,"journal":{"name":"Journal of hematology","volume":"12 1","pages":"49-58"},"PeriodicalIF":1.3000,"publicationDate":"2023-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3e/6e/jh-12-049.PMC9990712.pdf","citationCount":"0","resultStr":"{\"title\":\"Transformed Plasmablastic Lymphoma Presenting With Marked Lymphocytosis and Spontaneous Tumor Lysis Syndrome.\",\"authors\":\"Yannis Hadjiyannis, Cecelia Miller, Norris I Hollie, Jayalakshmi Balakrishna, Francesca Cottini\",\"doi\":\"10.14740/jh1067\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The clinicopathology entity of plasmablastic lymphoma (PBL), despite broad recognition by the World Health Organization (WHO), represents a diagnostic challenge due to its overlapping features and scarce occurrence. Often, PBL arises in immunodeficient, elderly male patients, most notably those who are human immunodeficiency virus (HIV)-positive. More infrequent, cases of transformed PBL (tPBL) evolved from another hematologic disease have been identified. Herein, we describe a case of a 65-year-old male transferred from a neighboring hospital with pronounced lymphocytosis and spontaneous tumor lysis syndrome (sTLS) presumed to be chronic lymphocytic leukemia (CLL). Utilizing a complete clinical, morphologic, immunophenotypic, and molecular evaluation, we arrived at a final diagnosis of tPBL with sTLS, suspected to have evolved from the <i>NF-κB/NOTCH/KLF2</i> (NNK) genetic cluster of splenic marginal zone lymphoma (SMZL) (NNK-SMZL), a potential transformation and presentation, to our knowledge, not previously reported. However, definitive clonality testing was not performed. In this report, we also outline the diagnostic and educational considerations we faced in discerning tPBL from other more common B-cell malignancies which can present similarly, such as CLL, mantle cell lymphoma, or plasmablastic myeloma. We summarize recently reported molecular, prognostic, and therapeutic considerations for the treatment and recognition of PBL, including the successful implementation, in our patient, of bortezomib to an EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) regimen with prophylactic intrathecal methotrexate, who has since achieved complete remission (CR) and entered clinical surveillance. Lastly, this report briefly highlights the challenge we faced in this area of hematologic typification that necessitates additional review and discussion by the WHO: tPBL with potential double-hit cytogenetic versus double-hit lymphoma with a plasmablastic phenotype.</p>\",\"PeriodicalId\":15964,\"journal\":{\"name\":\"Journal of hematology\",\"volume\":\"12 1\",\"pages\":\"49-58\"},\"PeriodicalIF\":1.3000,\"publicationDate\":\"2023-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/3e/6e/jh-12-049.PMC9990712.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of hematology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.14740/jh1067\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of hematology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.14740/jh1067","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Transformed Plasmablastic Lymphoma Presenting With Marked Lymphocytosis and Spontaneous Tumor Lysis Syndrome.
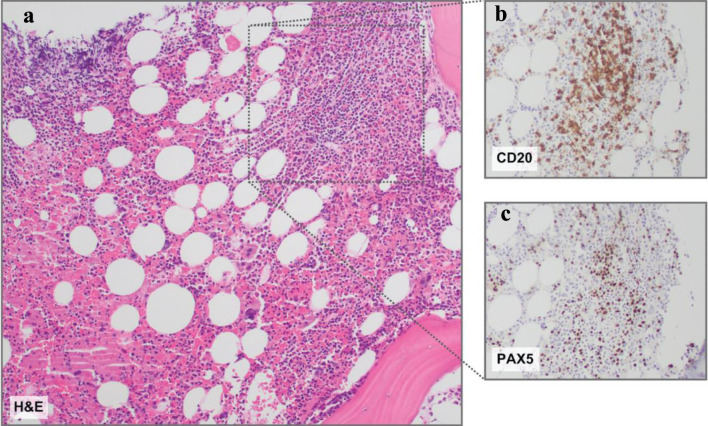
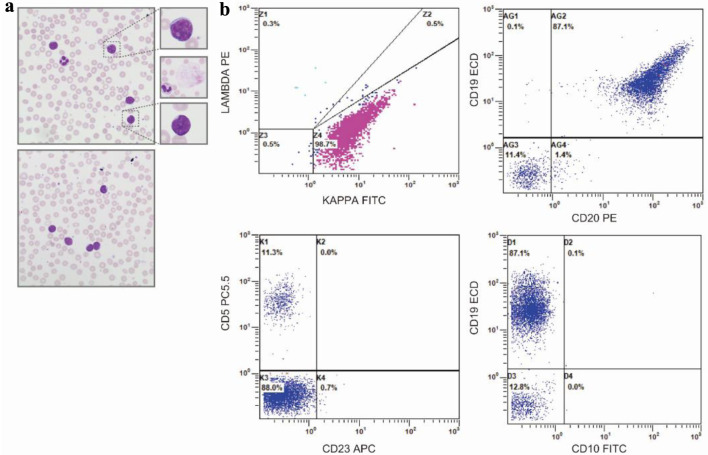
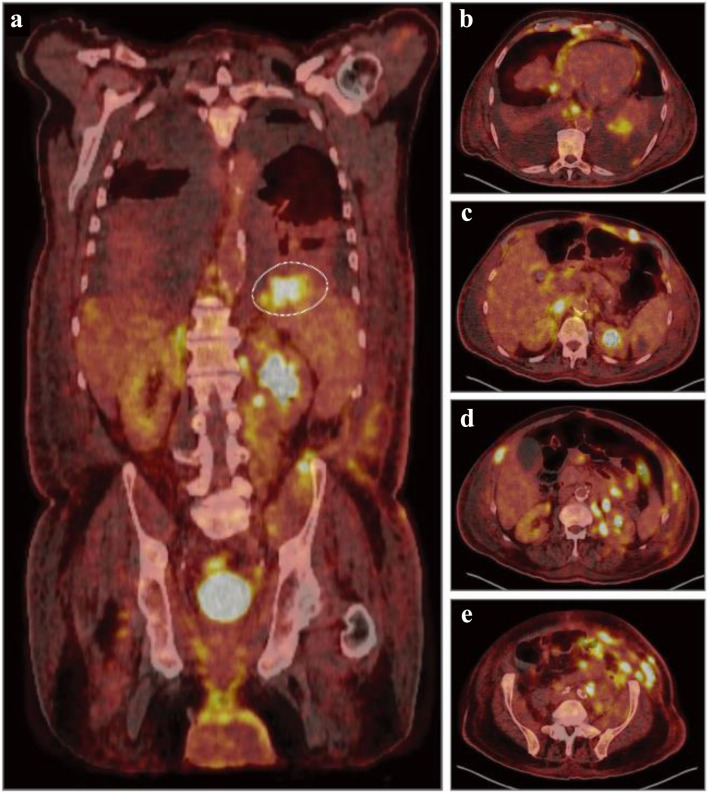
The clinicopathology entity of plasmablastic lymphoma (PBL), despite broad recognition by the World Health Organization (WHO), represents a diagnostic challenge due to its overlapping features and scarce occurrence. Often, PBL arises in immunodeficient, elderly male patients, most notably those who are human immunodeficiency virus (HIV)-positive. More infrequent, cases of transformed PBL (tPBL) evolved from another hematologic disease have been identified. Herein, we describe a case of a 65-year-old male transferred from a neighboring hospital with pronounced lymphocytosis and spontaneous tumor lysis syndrome (sTLS) presumed to be chronic lymphocytic leukemia (CLL). Utilizing a complete clinical, morphologic, immunophenotypic, and molecular evaluation, we arrived at a final diagnosis of tPBL with sTLS, suspected to have evolved from the NF-κB/NOTCH/KLF2 (NNK) genetic cluster of splenic marginal zone lymphoma (SMZL) (NNK-SMZL), a potential transformation and presentation, to our knowledge, not previously reported. However, definitive clonality testing was not performed. In this report, we also outline the diagnostic and educational considerations we faced in discerning tPBL from other more common B-cell malignancies which can present similarly, such as CLL, mantle cell lymphoma, or plasmablastic myeloma. We summarize recently reported molecular, prognostic, and therapeutic considerations for the treatment and recognition of PBL, including the successful implementation, in our patient, of bortezomib to an EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) regimen with prophylactic intrathecal methotrexate, who has since achieved complete remission (CR) and entered clinical surveillance. Lastly, this report briefly highlights the challenge we faced in this area of hematologic typification that necessitates additional review and discussion by the WHO: tPBL with potential double-hit cytogenetic versus double-hit lymphoma with a plasmablastic phenotype.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
