Hela Koka, Clara Bodelon, Steve Horvath, Priscilla Ming Yi Lee, Difei Wang, Lei Song, Tongwu Zhang, Amber N Hurson, Jennifer Lyn Guida, Bin Zhu, Maeve Bailey-Whyte, Feng Wang, Cherry Wu, Koon Ho Tsang, Yee-Kei Tsoi, W C Chan, Sze Hong Law, Ray Ka Wai Hung, Gary M Tse, Karen Ka-Wan Yuen, Eric Karlins, Kristine Jones, Aurelie Vogt, Bin Zhu, Amy Hutchinson, Belynda Hicks, Montserrat Garcia-Closas, Stephen Chanock, Jill Barnholtz-Sloan, Lap Ah Tse, Xiaohong R Yang
{"title":"中国女性乳腺癌配对肿瘤及邻近正常乳腺组织DNA甲基化年龄","authors":"Hela Koka, Clara Bodelon, Steve Horvath, Priscilla Ming Yi Lee, Difei Wang, Lei Song, Tongwu Zhang, Amber N Hurson, Jennifer Lyn Guida, Bin Zhu, Maeve Bailey-Whyte, Feng Wang, Cherry Wu, Koon Ho Tsang, Yee-Kei Tsoi, W C Chan, Sze Hong Law, Ray Ka Wai Hung, Gary M Tse, Karen Ka-Wan Yuen, Eric Karlins, Kristine Jones, Aurelie Vogt, Bin Zhu, Amy Hutchinson, Belynda Hicks, Montserrat Garcia-Closas, Stephen Chanock, Jill Barnholtz-Sloan, Lap Ah Tse, Xiaohong R Yang","doi":"10.1186/s13148-023-01465-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Few studies have examined epigenetic age acceleration (AA), the difference between DNA methylation (DNAm) predicted age and chronological age, in relation to somatic genomic features in paired cancer and normal tissue, with less work done in non-European populations. In this study, we aimed to examine DNAm age and its associations with breast cancer risk factors, subtypes, somatic genomic profiles including mutation and copy number alterations and other aging markers in breast tissue of Chinese breast cancer (BC) patients from Hong Kong.</p><p><strong>Methods: </strong>We performed genome-wide DNA methylation profiling of 196 tumor and 188 paired adjacent normal tissue collected from Chinese BC patients in Hong Kong (HKBC) using Illumina MethylationEPIC array. The DNAm age was calculated using Horvath's pan-tissue clock model. Somatic genomic features were based on data from RNA sequencing (RNASeq), whole-exome sequencing (WES), and whole-genome sequencing (WGS). Pearson's correlation (r), Kruskal-Wallis test, and regression models were used to estimate associations of DNAm AA with somatic features and breast cancer risk factors.</p><p><strong>Results: </strong>DNAm age showed a stronger correlation with chronological age in normal (Pearson r = 0.78, P < 2.2e-16) than in tumor tissue (Pearson r = 0.31, P = 7.8e-06). Although overall DNAm age or AA did not vary significantly by tissue within the same individual, luminal A tumors exhibited increased DNAm AA (P = 0.004) while HER2-enriched/basal-like tumors exhibited markedly lower DNAm AA (P = < .0001) compared with paired normal tissue. Consistent with the subtype association, tumor DNAm AA was positively correlated with ESR1 (Pearson r = 0.39, P = 6.3e-06) and PGR (Pearson r = 0.36, P = 2.4e-05) gene expression. In line with this, we found that increasing DNAm AA was associated with higher body mass index (P = 0.039) and earlier age at menarche (P = 0.035), factors that are related to cumulative exposure to estrogen. In contrast, variables indicating extensive genomic instability, such as TP53 somatic mutations, high tumor mutation/copy number alteration burden, and homologous repair deficiency were associated with lower DNAm AA.</p><p><strong>Conclusions: </strong>Our findings provide additional insights into the complexity of breast tissue aging that is associated with the interaction of hormonal, genomic, and epigenetic mechanisms in an East Asian population.</p>","PeriodicalId":48652,"journal":{"name":"Clinical Epigenetics","volume":"15 1","pages":"55"},"PeriodicalIF":4.4000,"publicationDate":"2023-03-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10062015/pdf/","citationCount":"0","resultStr":"{\"title\":\"DNA methylation age in paired tumor and adjacent normal breast tissue in Chinese women with breast cancer.\",\"authors\":\"Hela Koka, Clara Bodelon, Steve Horvath, Priscilla Ming Yi Lee, Difei Wang, Lei Song, Tongwu Zhang, Amber N Hurson, Jennifer Lyn Guida, Bin Zhu, Maeve Bailey-Whyte, Feng Wang, Cherry Wu, Koon Ho Tsang, Yee-Kei Tsoi, W C Chan, Sze Hong Law, Ray Ka Wai Hung, Gary M Tse, Karen Ka-Wan Yuen, Eric Karlins, Kristine Jones, Aurelie Vogt, Bin Zhu, Amy Hutchinson, Belynda Hicks, Montserrat Garcia-Closas, Stephen Chanock, Jill Barnholtz-Sloan, Lap Ah Tse, Xiaohong R Yang\",\"doi\":\"10.1186/s13148-023-01465-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Few studies have examined epigenetic age acceleration (AA), the difference between DNA methylation (DNAm) predicted age and chronological age, in relation to somatic genomic features in paired cancer and normal tissue, with less work done in non-European populations. In this study, we aimed to examine DNAm age and its associations with breast cancer risk factors, subtypes, somatic genomic profiles including mutation and copy number alterations and other aging markers in breast tissue of Chinese breast cancer (BC) patients from Hong Kong.</p><p><strong>Methods: </strong>We performed genome-wide DNA methylation profiling of 196 tumor and 188 paired adjacent normal tissue collected from Chinese BC patients in Hong Kong (HKBC) using Illumina MethylationEPIC array. The DNAm age was calculated using Horvath's pan-tissue clock model. Somatic genomic features were based on data from RNA sequencing (RNASeq), whole-exome sequencing (WES), and whole-genome sequencing (WGS). Pearson's correlation (r), Kruskal-Wallis test, and regression models were used to estimate associations of DNAm AA with somatic features and breast cancer risk factors.</p><p><strong>Results: </strong>DNAm age showed a stronger correlation with chronological age in normal (Pearson r = 0.78, P < 2.2e-16) than in tumor tissue (Pearson r = 0.31, P = 7.8e-06). Although overall DNAm age or AA did not vary significantly by tissue within the same individual, luminal A tumors exhibited increased DNAm AA (P = 0.004) while HER2-enriched/basal-like tumors exhibited markedly lower DNAm AA (P = < .0001) compared with paired normal tissue. Consistent with the subtype association, tumor DNAm AA was positively correlated with ESR1 (Pearson r = 0.39, P = 6.3e-06) and PGR (Pearson r = 0.36, P = 2.4e-05) gene expression. In line with this, we found that increasing DNAm AA was associated with higher body mass index (P = 0.039) and earlier age at menarche (P = 0.035), factors that are related to cumulative exposure to estrogen. In contrast, variables indicating extensive genomic instability, such as TP53 somatic mutations, high tumor mutation/copy number alteration burden, and homologous repair deficiency were associated with lower DNAm AA.</p><p><strong>Conclusions: </strong>Our findings provide additional insights into the complexity of breast tissue aging that is associated with the interaction of hormonal, genomic, and epigenetic mechanisms in an East Asian population.</p>\",\"PeriodicalId\":48652,\"journal\":{\"name\":\"Clinical Epigenetics\",\"volume\":\"15 1\",\"pages\":\"55\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2023-03-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10062015/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Epigenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13148-023-01465-1\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Epigenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13148-023-01465-1","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
DNA methylation age in paired tumor and adjacent normal breast tissue in Chinese women with breast cancer.
Background: Few studies have examined epigenetic age acceleration (AA), the difference between DNA methylation (DNAm) predicted age and chronological age, in relation to somatic genomic features in paired cancer and normal tissue, with less work done in non-European populations. In this study, we aimed to examine DNAm age and its associations with breast cancer risk factors, subtypes, somatic genomic profiles including mutation and copy number alterations and other aging markers in breast tissue of Chinese breast cancer (BC) patients from Hong Kong.
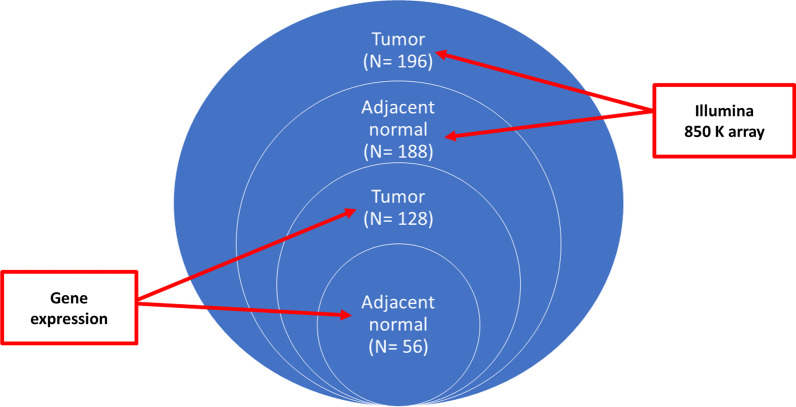
Methods: We performed genome-wide DNA methylation profiling of 196 tumor and 188 paired adjacent normal tissue collected from Chinese BC patients in Hong Kong (HKBC) using Illumina MethylationEPIC array. The DNAm age was calculated using Horvath's pan-tissue clock model. Somatic genomic features were based on data from RNA sequencing (RNASeq), whole-exome sequencing (WES), and whole-genome sequencing (WGS). Pearson's correlation (r), Kruskal-Wallis test, and regression models were used to estimate associations of DNAm AA with somatic features and breast cancer risk factors.
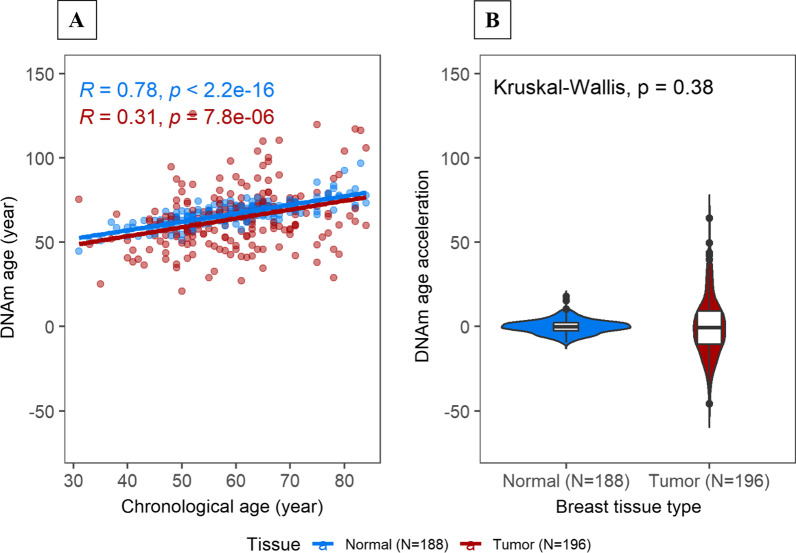
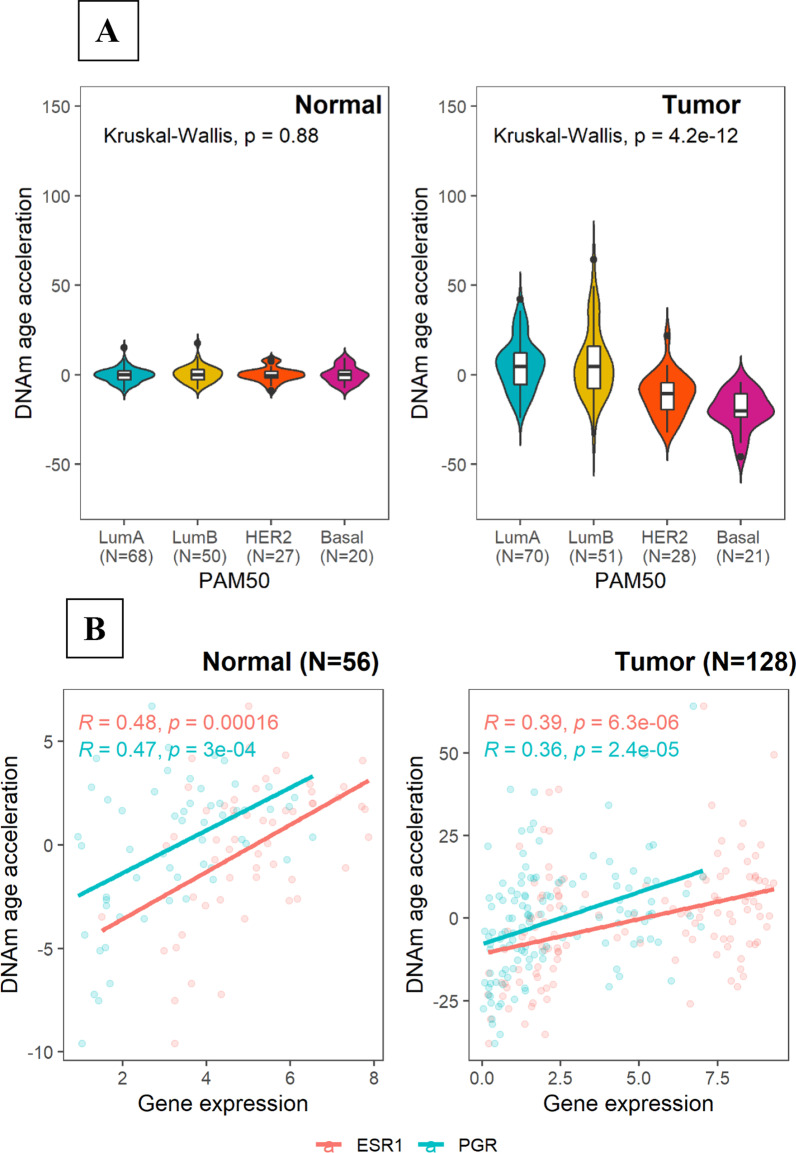
Results: DNAm age showed a stronger correlation with chronological age in normal (Pearson r = 0.78, P < 2.2e-16) than in tumor tissue (Pearson r = 0.31, P = 7.8e-06). Although overall DNAm age or AA did not vary significantly by tissue within the same individual, luminal A tumors exhibited increased DNAm AA (P = 0.004) while HER2-enriched/basal-like tumors exhibited markedly lower DNAm AA (P = < .0001) compared with paired normal tissue. Consistent with the subtype association, tumor DNAm AA was positively correlated with ESR1 (Pearson r = 0.39, P = 6.3e-06) and PGR (Pearson r = 0.36, P = 2.4e-05) gene expression. In line with this, we found that increasing DNAm AA was associated with higher body mass index (P = 0.039) and earlier age at menarche (P = 0.035), factors that are related to cumulative exposure to estrogen. In contrast, variables indicating extensive genomic instability, such as TP53 somatic mutations, high tumor mutation/copy number alteration burden, and homologous repair deficiency were associated with lower DNAm AA.
Conclusions: Our findings provide additional insights into the complexity of breast tissue aging that is associated with the interaction of hormonal, genomic, and epigenetic mechanisms in an East Asian population.
Clinical EpigeneticsBiochemistry, Genetics and Molecular Biology-Developmental Biology
CiteScore
8.90
自引率
5.30%
发文量
150
审稿时长
12 weeks
期刊介绍:
Clinical Epigenetics, the official journal of the Clinical Epigenetics Society, is an open access, peer-reviewed journal that encompasses all aspects of epigenetic principles and mechanisms in relation to human disease, diagnosis and therapy. Clinical trials and research in disease model organisms are particularly welcome.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
