Divya S Bhat, Eva Malacaria, Ludovica Di Biagi, Mortezaali Razzaghi, Masayoshi Honda, Kathryn F Hobbs, Sarah R Hengel, Pietro Pichierri, M Ashley Spies, Maria Spies
{"title":"通过新型药物抑制剂对 RAD52-ssDNA 复合物进行治疗性破坏。","authors":"Divya S Bhat, Eva Malacaria, Ludovica Di Biagi, Mortezaali Razzaghi, Masayoshi Honda, Kathryn F Hobbs, Sarah R Hengel, Pietro Pichierri, M Ashley Spies, Maria Spies","doi":"10.1093/narcan/zcad018","DOIUrl":null,"url":null,"abstract":"<p><p>RAD52 protein is a coveted target for anticancer drug discovery. Similar to poly-ADP-ribose polymerase (PARP) inhibitors, pharmacological inhibition of RAD52 is synthetically lethal with defects in genome caretakers BRCA1 and BRCA2 (∼25% of breast and ovarian cancers). Emerging structure activity relationships for RAD52 are complex, making it challenging to transform previously identified disruptors of the RAD52-ssDNA interaction into drug-like leads using traditional medicinal chemistry approaches. Using pharmacophoric informatics on the RAD52 complexation by epigallocatechin (EGC), and the Enamine <i>in silico</i> REAL database, we identified six distinct chemical scaffolds that occupy the same physical space on RAD52 as EGC. All six were RAD52 inhibitors (IC<sub>50</sub> ∼23-1200 μM) with two of the compounds (Z56 and Z99) selectively killing BRCA-mutant cells and inhibiting cellular activities of RAD52 at micromolar inhibitor concentrations. While Z56 had no effect on the ssDNA-binding protein RPA and was toxic to BRCA-mutant cells only, Z99 inhibited both proteins and displayed toxicity towards BRCA-complemented cells. Optimization of the Z99 scaffold resulted in a set of more powerful and selective inhibitors (IC<sub>50</sub> ∼1.3-8 μM), which were only toxic to BRCA-mutant cells. RAD52 complexation by Z56, Z99 and its more specific derivatives provide a roadmap for next generation of cancer therapeutics.</p>","PeriodicalId":18879,"journal":{"name":"NAR Cancer","volume":"5 2","pages":"zcad018"},"PeriodicalIF":0.0000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/4d/zcad018.PMC10150327.pdf","citationCount":"0","resultStr":"{\"title\":\"Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors.\",\"authors\":\"Divya S Bhat, Eva Malacaria, Ludovica Di Biagi, Mortezaali Razzaghi, Masayoshi Honda, Kathryn F Hobbs, Sarah R Hengel, Pietro Pichierri, M Ashley Spies, Maria Spies\",\"doi\":\"10.1093/narcan/zcad018\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>RAD52 protein is a coveted target for anticancer drug discovery. Similar to poly-ADP-ribose polymerase (PARP) inhibitors, pharmacological inhibition of RAD52 is synthetically lethal with defects in genome caretakers BRCA1 and BRCA2 (∼25% of breast and ovarian cancers). Emerging structure activity relationships for RAD52 are complex, making it challenging to transform previously identified disruptors of the RAD52-ssDNA interaction into drug-like leads using traditional medicinal chemistry approaches. Using pharmacophoric informatics on the RAD52 complexation by epigallocatechin (EGC), and the Enamine <i>in silico</i> REAL database, we identified six distinct chemical scaffolds that occupy the same physical space on RAD52 as EGC. All six were RAD52 inhibitors (IC<sub>50</sub> ∼23-1200 μM) with two of the compounds (Z56 and Z99) selectively killing BRCA-mutant cells and inhibiting cellular activities of RAD52 at micromolar inhibitor concentrations. While Z56 had no effect on the ssDNA-binding protein RPA and was toxic to BRCA-mutant cells only, Z99 inhibited both proteins and displayed toxicity towards BRCA-complemented cells. Optimization of the Z99 scaffold resulted in a set of more powerful and selective inhibitors (IC<sub>50</sub> ∼1.3-8 μM), which were only toxic to BRCA-mutant cells. RAD52 complexation by Z56, Z99 and its more specific derivatives provide a roadmap for next generation of cancer therapeutics.</p>\",\"PeriodicalId\":18879,\"journal\":{\"name\":\"NAR Cancer\",\"volume\":\"5 2\",\"pages\":\"zcad018\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/1c/4d/zcad018.PMC10150327.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NAR Cancer\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/narcan/zcad018\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/6/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/narcan/zcad018","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/6/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Therapeutic disruption of RAD52-ssDNA complexation via novel drug-like inhibitors.
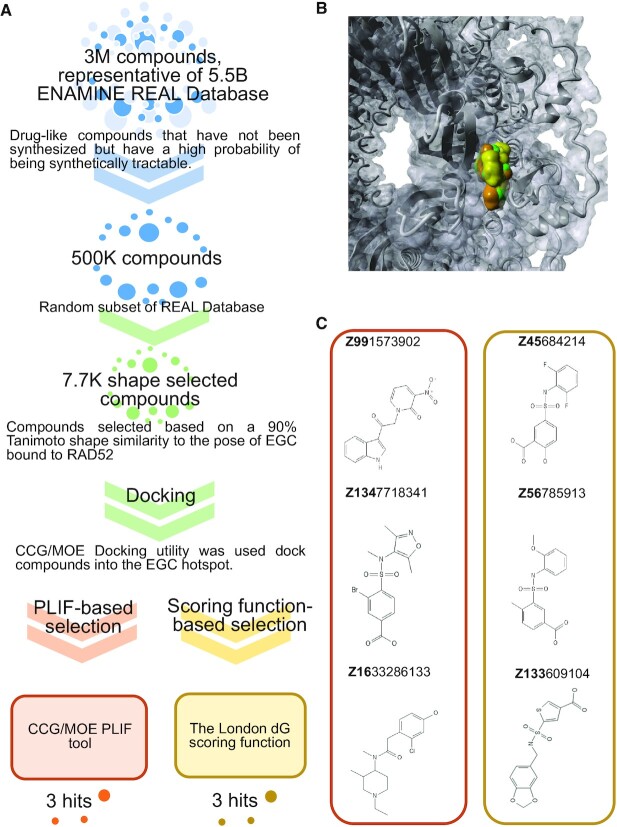
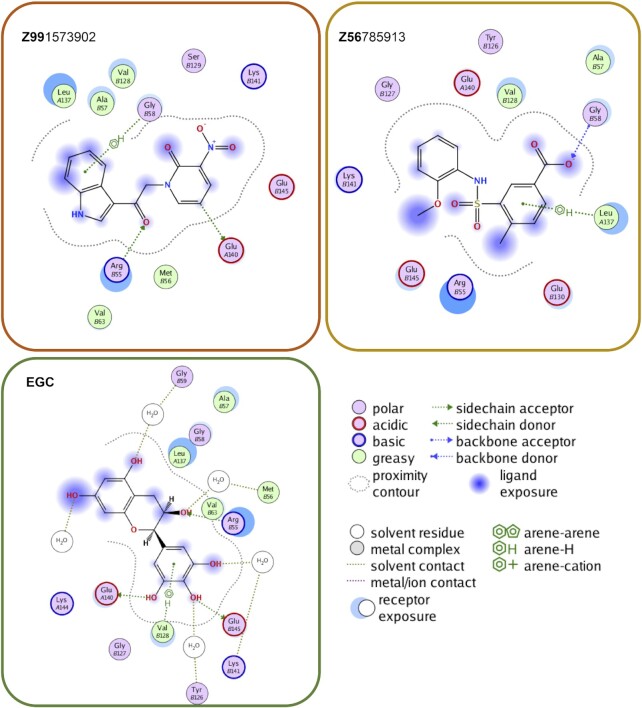
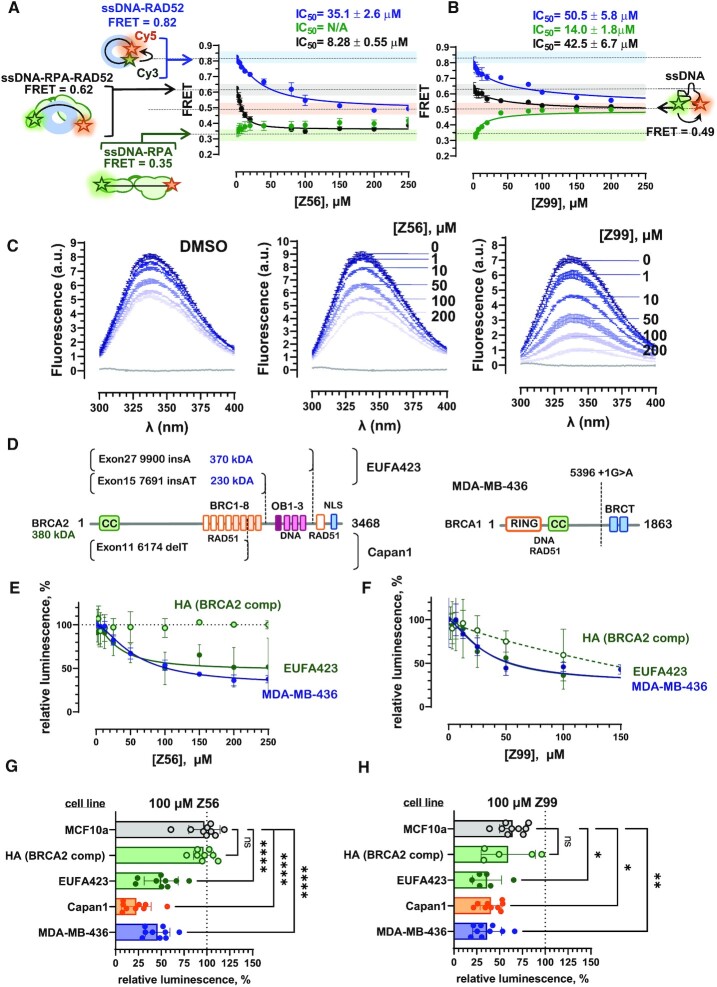
RAD52 protein is a coveted target for anticancer drug discovery. Similar to poly-ADP-ribose polymerase (PARP) inhibitors, pharmacological inhibition of RAD52 is synthetically lethal with defects in genome caretakers BRCA1 and BRCA2 (∼25% of breast and ovarian cancers). Emerging structure activity relationships for RAD52 are complex, making it challenging to transform previously identified disruptors of the RAD52-ssDNA interaction into drug-like leads using traditional medicinal chemistry approaches. Using pharmacophoric informatics on the RAD52 complexation by epigallocatechin (EGC), and the Enamine in silico REAL database, we identified six distinct chemical scaffolds that occupy the same physical space on RAD52 as EGC. All six were RAD52 inhibitors (IC50 ∼23-1200 μM) with two of the compounds (Z56 and Z99) selectively killing BRCA-mutant cells and inhibiting cellular activities of RAD52 at micromolar inhibitor concentrations. While Z56 had no effect on the ssDNA-binding protein RPA and was toxic to BRCA-mutant cells only, Z99 inhibited both proteins and displayed toxicity towards BRCA-complemented cells. Optimization of the Z99 scaffold resulted in a set of more powerful and selective inhibitors (IC50 ∼1.3-8 μM), which were only toxic to BRCA-mutant cells. RAD52 complexation by Z56, Z99 and its more specific derivatives provide a roadmap for next generation of cancer therapeutics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
