Ling-Yu Wang, Lu-Qiang Zhang, Qian-Zhong Li, Hui Bai
{"title":"H3K36me3和H3K79me2调控基因在乳腺癌中的风险模型构建。","authors":"Ling-Yu Wang, Lu-Qiang Zhang, Qian-Zhong Li, Hui Bai","doi":"10.52601/bpr.2023.220022","DOIUrl":null,"url":null,"abstract":"<p><p>Abnormal histone modifications (HMs) can promote the occurrence of breast cancer. To elucidate the relationship between HMs and gene expression, we analyzed HM binding patterns and calculated their signal changes between breast tumor cells and normal cells. On this basis, the influences of HM signal changes on the expression changes of breast cancer-related genes were estimated by three different methods. The results showed that H3K79me2 and H3K36me3 may contribute more to gene expression changes. Subsequently, 2109 genes with differential H3K79me2 or H3K36me3 levels during cancerogenesis were identified by the Shannon entropy and submitted to perform functional enrichment analyses. Enrichment analyses displayed that these genes were involved in pathways in cancer, human papillomavirus infection, and viral carcinogenesis. Univariate Cox, LASSO, and multivariate Cox regression analyses were then adopted, and nine potential breast cancer-related driver genes were extracted from the genes with differential H3K79me2/H3K36me3 levels in the TCGA cohort. To facilitate the application, the expression levels of nine driver genes were transformed into a risk score model, and its robustness was tested via time-dependent receiver operating characteristic curves in the TCGA dataset and an independent GEO dataset. At last, the distribution levels of H3K79me2 and H3K36me3 in the nine driver genes were reanalyzed in the two cell lines and the regions with significant signal changes were located.</p>","PeriodicalId":59621,"journal":{"name":"生物物理学报:英文版","volume":"9 1","pages":"45-56"},"PeriodicalIF":0.0000,"publicationDate":"2023-02-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10323774/pdf/","citationCount":"0","resultStr":"{\"title\":\"The risk model construction of the genes regulated by H3K36me3 and H3K79me2 in breast cancer.\",\"authors\":\"Ling-Yu Wang, Lu-Qiang Zhang, Qian-Zhong Li, Hui Bai\",\"doi\":\"10.52601/bpr.2023.220022\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Abnormal histone modifications (HMs) can promote the occurrence of breast cancer. To elucidate the relationship between HMs and gene expression, we analyzed HM binding patterns and calculated their signal changes between breast tumor cells and normal cells. On this basis, the influences of HM signal changes on the expression changes of breast cancer-related genes were estimated by three different methods. The results showed that H3K79me2 and H3K36me3 may contribute more to gene expression changes. Subsequently, 2109 genes with differential H3K79me2 or H3K36me3 levels during cancerogenesis were identified by the Shannon entropy and submitted to perform functional enrichment analyses. Enrichment analyses displayed that these genes were involved in pathways in cancer, human papillomavirus infection, and viral carcinogenesis. Univariate Cox, LASSO, and multivariate Cox regression analyses were then adopted, and nine potential breast cancer-related driver genes were extracted from the genes with differential H3K79me2/H3K36me3 levels in the TCGA cohort. To facilitate the application, the expression levels of nine driver genes were transformed into a risk score model, and its robustness was tested via time-dependent receiver operating characteristic curves in the TCGA dataset and an independent GEO dataset. At last, the distribution levels of H3K79me2 and H3K36me3 in the nine driver genes were reanalyzed in the two cell lines and the regions with significant signal changes were located.</p>\",\"PeriodicalId\":59621,\"journal\":{\"name\":\"生物物理学报:英文版\",\"volume\":\"9 1\",\"pages\":\"45-56\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2023-02-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10323774/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"生物物理学报:英文版\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.52601/bpr.2023.220022\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"生物物理学报:英文版","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.52601/bpr.2023.220022","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
The risk model construction of the genes regulated by H3K36me3 and H3K79me2 in breast cancer.
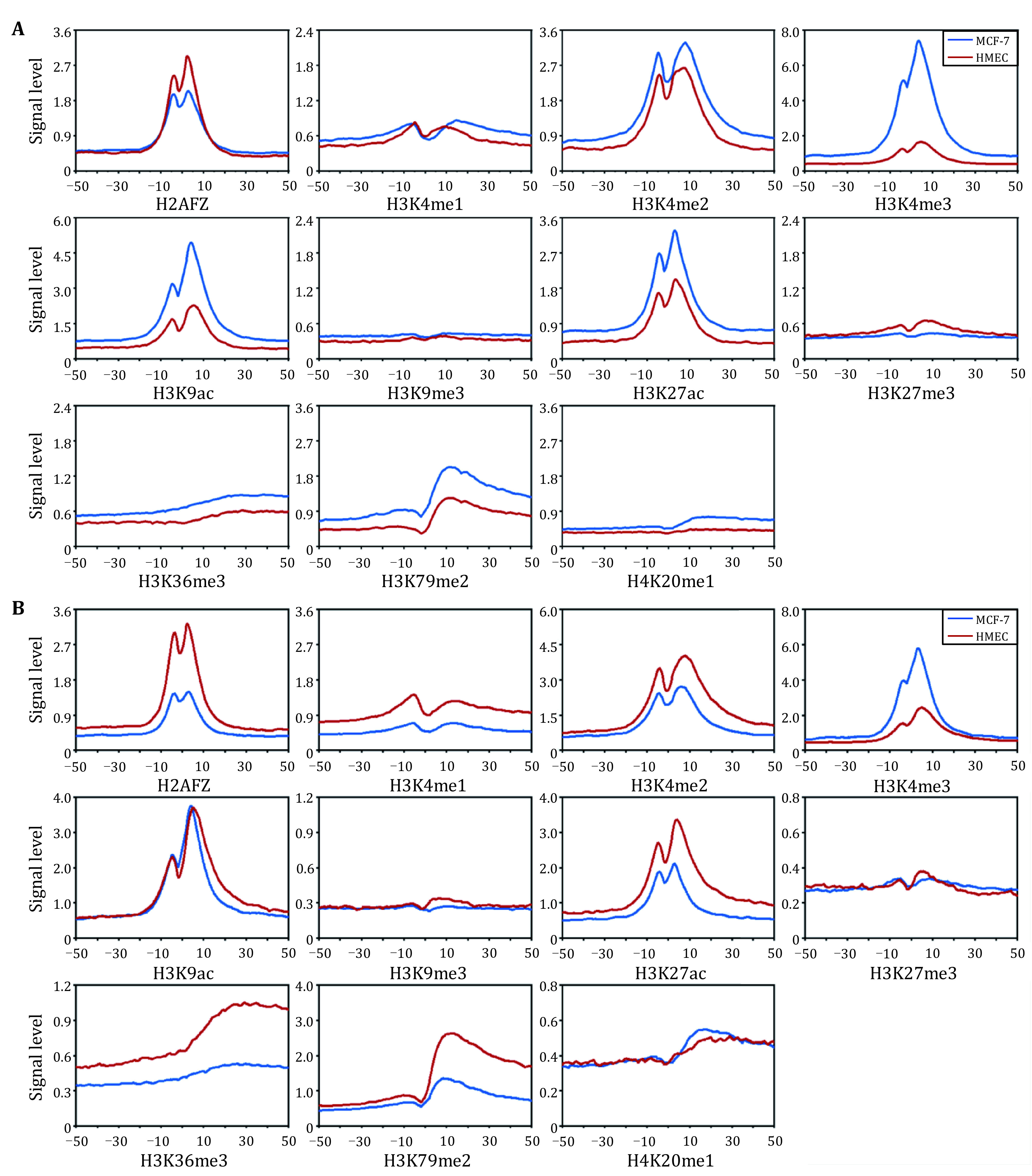
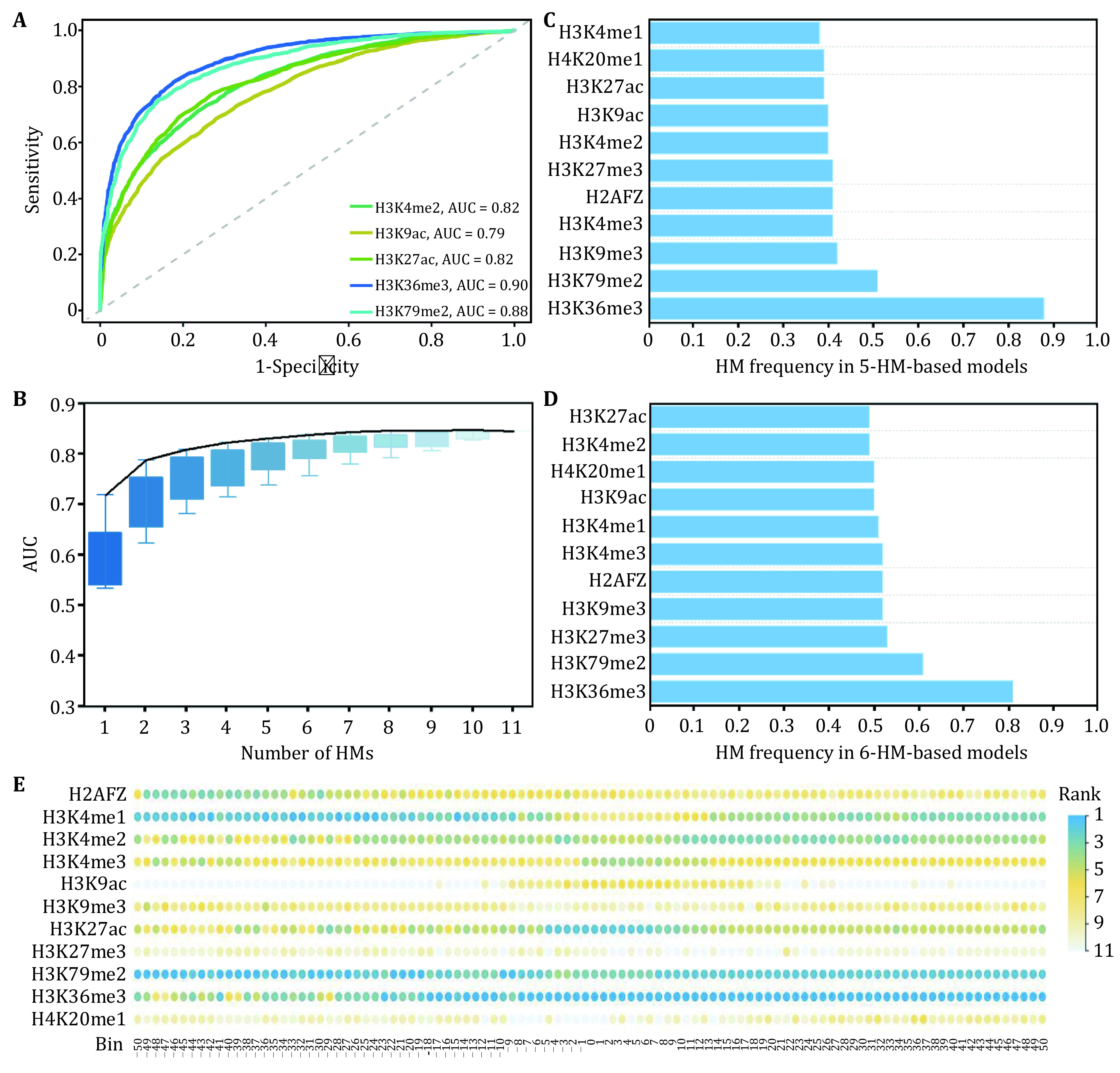
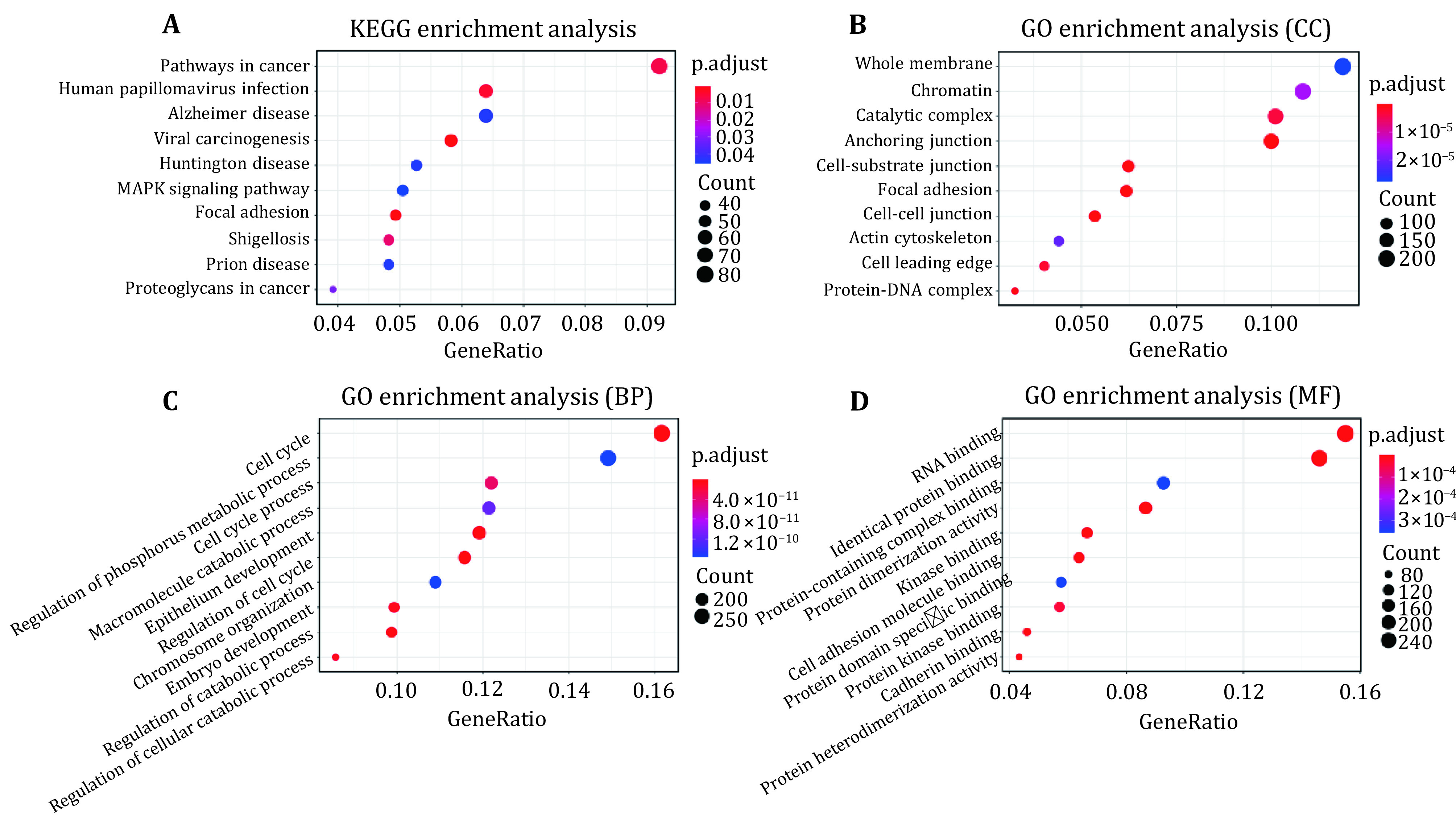
Abnormal histone modifications (HMs) can promote the occurrence of breast cancer. To elucidate the relationship between HMs and gene expression, we analyzed HM binding patterns and calculated their signal changes between breast tumor cells and normal cells. On this basis, the influences of HM signal changes on the expression changes of breast cancer-related genes were estimated by three different methods. The results showed that H3K79me2 and H3K36me3 may contribute more to gene expression changes. Subsequently, 2109 genes with differential H3K79me2 or H3K36me3 levels during cancerogenesis were identified by the Shannon entropy and submitted to perform functional enrichment analyses. Enrichment analyses displayed that these genes were involved in pathways in cancer, human papillomavirus infection, and viral carcinogenesis. Univariate Cox, LASSO, and multivariate Cox regression analyses were then adopted, and nine potential breast cancer-related driver genes were extracted from the genes with differential H3K79me2/H3K36me3 levels in the TCGA cohort. To facilitate the application, the expression levels of nine driver genes were transformed into a risk score model, and its robustness was tested via time-dependent receiver operating characteristic curves in the TCGA dataset and an independent GEO dataset. At last, the distribution levels of H3K79me2 and H3K36me3 in the nine driver genes were reanalyzed in the two cell lines and the regions with significant signal changes were located.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
