{"title":"青少年发病原发性辅酶Q10缺乏伴罕见的辅酶q8a基因突变:1例报告及文献复习。","authors":"Mahsa Hojabri, Abolfazl Gilani, Rana Irilouzadian, Habibe Nejad Biglari, Roham Sarmadian","doi":"10.1177/11795476231188061","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary deficiency of coenzyme Q<sub>10</sub> deficiency-4 (CoQ<sub>10</sub>D4) is a heterogeneous disorder affecting different age groups. The main clinical manifestation consists of cerebellar ataxia, exercise intolerance, and dystonia.</p><p><strong>Case report: </strong>We provide a case of adolescence-onset ataxia, head tremor, and proximal muscle weakness accompanied by psychiatric features and abnormal serum urea (49.4 mg/dL), lactate (7.5 mmol/L), and CoQ10 level (0.4 µg/mL). Brain-MRI demonstrated cerebellar atrophy, thinning of the corpus callosum, and loss of white matter. Whole exome sequencing showed a homozygous missense mutation (c.911C>T; p.A304V) in CoQ8A gene which is a rare mutation and responsible variant of CoQ<sub>10</sub>D4. After supplementary treatment with CoQ<sub>10</sub> 50 mg/twice a day for 2 months the clinical symptoms improved.</p><p><strong>Conclusion: </strong>These observations highlight the significance of the early diagnosis of potentially treatable CoQ8A mutation as well as patient education and follow-up. Our findings widen the spectrum of CoQ8A phenotypic features so that clinicians be familiar with the disease not only in severe childhood-onset ataxia but also in adolescence with accompanying psychiatric problems.</p>","PeriodicalId":10357,"journal":{"name":"Clinical Medicine Insights. Case Reports","volume":"16 ","pages":"11795476231188061"},"PeriodicalIF":0.8000,"publicationDate":"2023-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e0/0d/10.1177_11795476231188061.PMC10354825.pdf","citationCount":"0","resultStr":"{\"title\":\"Adolescence Onset Primary Coenzyme Q10 Deficiency With Rare CoQ8A Gene Mutation: A Case Report and Review of Literature.\",\"authors\":\"Mahsa Hojabri, Abolfazl Gilani, Rana Irilouzadian, Habibe Nejad Biglari, Roham Sarmadian\",\"doi\":\"10.1177/11795476231188061\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Primary deficiency of coenzyme Q<sub>10</sub> deficiency-4 (CoQ<sub>10</sub>D4) is a heterogeneous disorder affecting different age groups. The main clinical manifestation consists of cerebellar ataxia, exercise intolerance, and dystonia.</p><p><strong>Case report: </strong>We provide a case of adolescence-onset ataxia, head tremor, and proximal muscle weakness accompanied by psychiatric features and abnormal serum urea (49.4 mg/dL), lactate (7.5 mmol/L), and CoQ10 level (0.4 µg/mL). Brain-MRI demonstrated cerebellar atrophy, thinning of the corpus callosum, and loss of white matter. Whole exome sequencing showed a homozygous missense mutation (c.911C>T; p.A304V) in CoQ8A gene which is a rare mutation and responsible variant of CoQ<sub>10</sub>D4. After supplementary treatment with CoQ<sub>10</sub> 50 mg/twice a day for 2 months the clinical symptoms improved.</p><p><strong>Conclusion: </strong>These observations highlight the significance of the early diagnosis of potentially treatable CoQ8A mutation as well as patient education and follow-up. Our findings widen the spectrum of CoQ8A phenotypic features so that clinicians be familiar with the disease not only in severe childhood-onset ataxia but also in adolescence with accompanying psychiatric problems.</p>\",\"PeriodicalId\":10357,\"journal\":{\"name\":\"Clinical Medicine Insights. Case Reports\",\"volume\":\"16 \",\"pages\":\"11795476231188061\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2023-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e0/0d/10.1177_11795476231188061.PMC10354825.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Medicine Insights. Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11795476231188061\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Medicine Insights. Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11795476231188061","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Adolescence Onset Primary Coenzyme Q10 Deficiency With Rare CoQ8A Gene Mutation: A Case Report and Review of Literature.
Background: Primary deficiency of coenzyme Q10 deficiency-4 (CoQ10D4) is a heterogeneous disorder affecting different age groups. The main clinical manifestation consists of cerebellar ataxia, exercise intolerance, and dystonia.
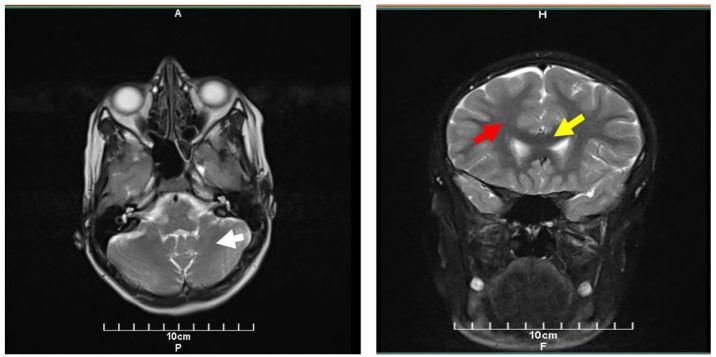
Case report: We provide a case of adolescence-onset ataxia, head tremor, and proximal muscle weakness accompanied by psychiatric features and abnormal serum urea (49.4 mg/dL), lactate (7.5 mmol/L), and CoQ10 level (0.4 µg/mL). Brain-MRI demonstrated cerebellar atrophy, thinning of the corpus callosum, and loss of white matter. Whole exome sequencing showed a homozygous missense mutation (c.911C>T; p.A304V) in CoQ8A gene which is a rare mutation and responsible variant of CoQ10D4. After supplementary treatment with CoQ10 50 mg/twice a day for 2 months the clinical symptoms improved.
Conclusion: These observations highlight the significance of the early diagnosis of potentially treatable CoQ8A mutation as well as patient education and follow-up. Our findings widen the spectrum of CoQ8A phenotypic features so that clinicians be familiar with the disease not only in severe childhood-onset ataxia but also in adolescence with accompanying psychiatric problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
