{"title":"DIA-MS2pep:从数据无关的采集数据中进行全面肽鉴定的无库框架。","authors":"Junjie Hou, Jifeng Wang, Fuquan Yang, Tao Xu","doi":"10.52601/bpr.2022.220011","DOIUrl":null,"url":null,"abstract":"<p><p>Identifying peptides directly from data-independent acquisition (DIA) data remains challenging due to the highly multiplexed MS/MS spectra. Spectral library-based peptide detection is sensitive, but it is limited to the depth of the library and mutes the discovery potential of DIA data. We present here, DIA-MS2pep, a library-free framework for comprehensive peptide identification from DIA data. DIA-MS2pep uses a data-driven algorithm for MS/MS spectrum demultiplexing using the fragments data without the need of a precursor. With a large precursor mass tolerance database search, DIA-MS2pep can identify the peptides and their modified forms. We demonstrate the performance of DIA-MS2pep by comparing it to conventional library-free tools in accuracy and sensitivity of peptide identifications using publicly available DIA datasets of varying samples, including HeLa cell lysates, phosphopeptides, plasma, <i>etc</i>. Compared with data-dependent acquisition-based spectral libraries, spectral libraries built directly from DIA data with DIA-MS2pep improve the accuracy and reproducibility of the quantitative proteome.</p>","PeriodicalId":59621,"journal":{"name":"生物物理学报:英文版","volume":"8 5-6","pages":"253-268"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10166510/pdf/","citationCount":"0","resultStr":"{\"title\":\"DIA-MS2pep: a library-free framework for comprehensive peptide identification from data-independent acquisition data.\",\"authors\":\"Junjie Hou, Jifeng Wang, Fuquan Yang, Tao Xu\",\"doi\":\"10.52601/bpr.2022.220011\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Identifying peptides directly from data-independent acquisition (DIA) data remains challenging due to the highly multiplexed MS/MS spectra. Spectral library-based peptide detection is sensitive, but it is limited to the depth of the library and mutes the discovery potential of DIA data. We present here, DIA-MS2pep, a library-free framework for comprehensive peptide identification from DIA data. DIA-MS2pep uses a data-driven algorithm for MS/MS spectrum demultiplexing using the fragments data without the need of a precursor. With a large precursor mass tolerance database search, DIA-MS2pep can identify the peptides and their modified forms. We demonstrate the performance of DIA-MS2pep by comparing it to conventional library-free tools in accuracy and sensitivity of peptide identifications using publicly available DIA datasets of varying samples, including HeLa cell lysates, phosphopeptides, plasma, <i>etc</i>. Compared with data-dependent acquisition-based spectral libraries, spectral libraries built directly from DIA data with DIA-MS2pep improve the accuracy and reproducibility of the quantitative proteome.</p>\",\"PeriodicalId\":59621,\"journal\":{\"name\":\"生物物理学报:英文版\",\"volume\":\"8 5-6\",\"pages\":\"253-268\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2022-12-31\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10166510/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"生物物理学报:英文版\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.52601/bpr.2022.220011\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"生物物理学报:英文版","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.52601/bpr.2022.220011","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
DIA-MS2pep: a library-free framework for comprehensive peptide identification from data-independent acquisition data.
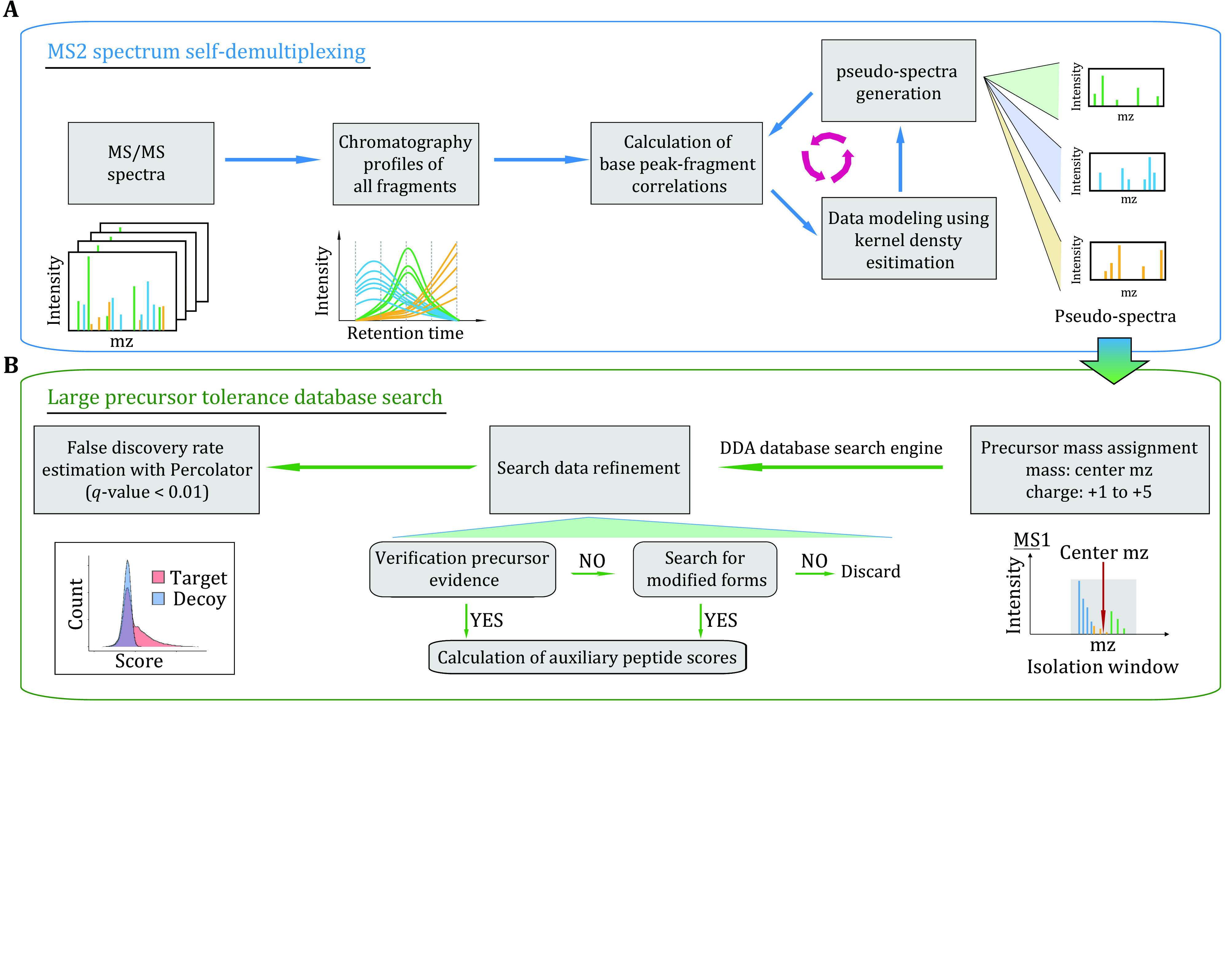
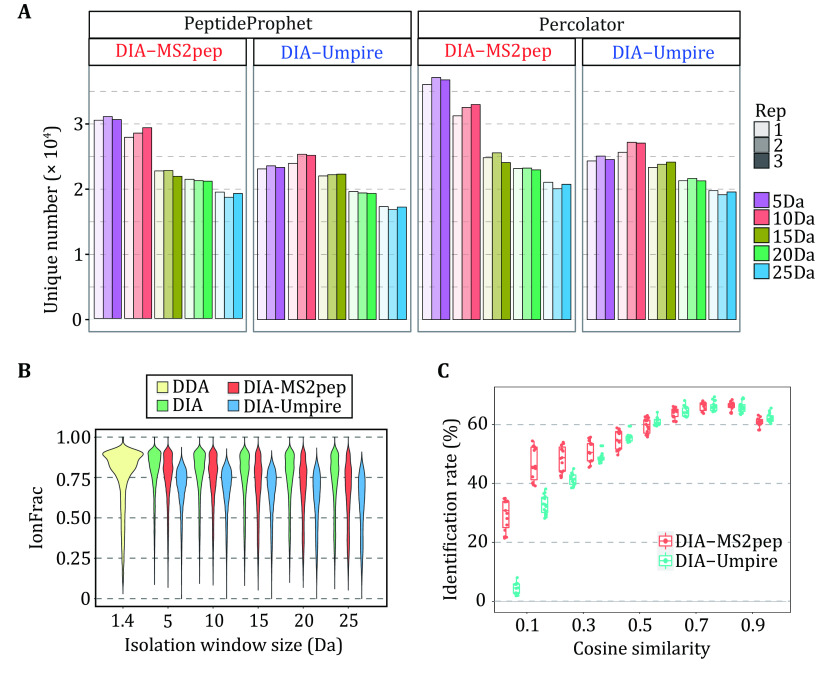
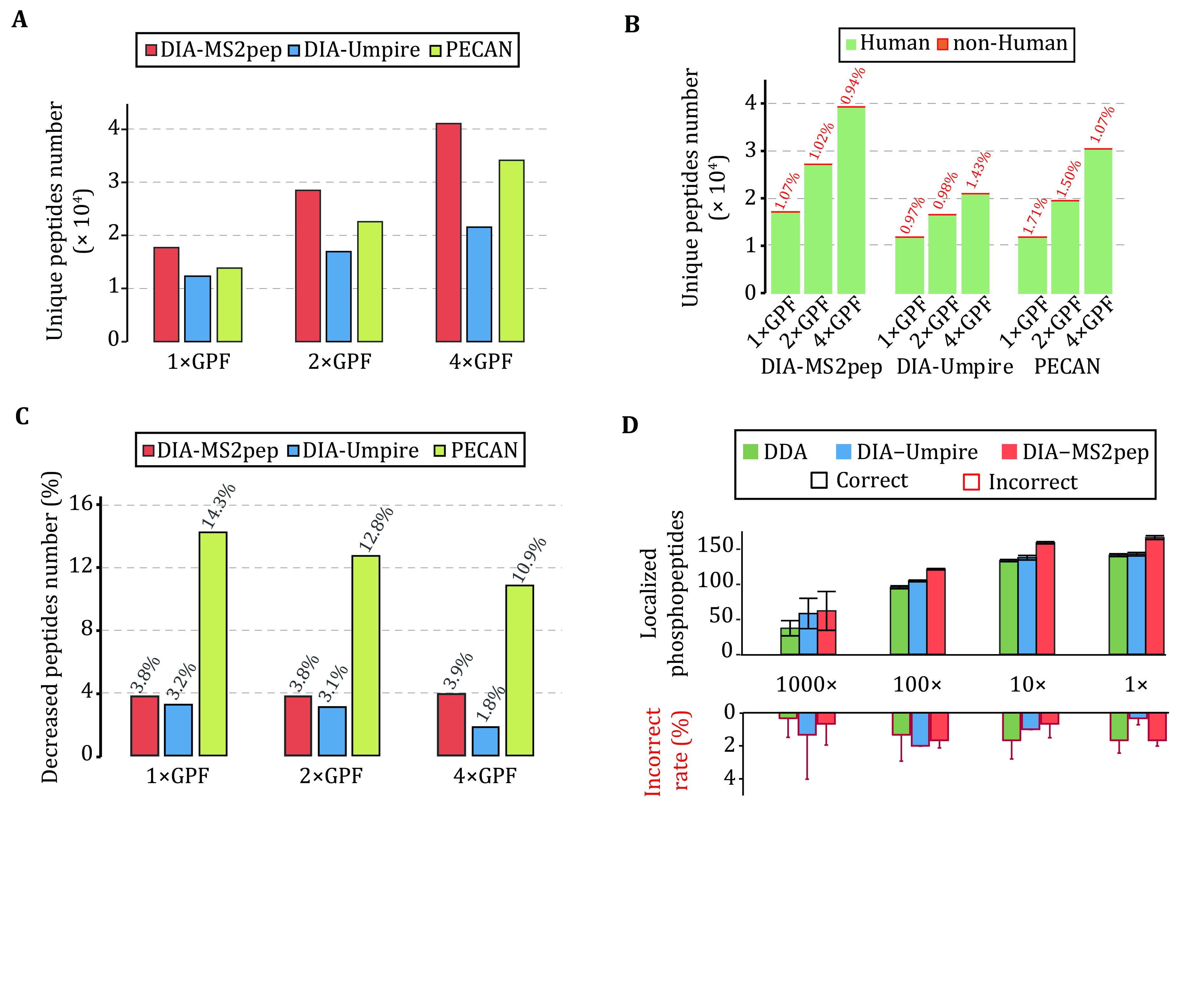
Identifying peptides directly from data-independent acquisition (DIA) data remains challenging due to the highly multiplexed MS/MS spectra. Spectral library-based peptide detection is sensitive, but it is limited to the depth of the library and mutes the discovery potential of DIA data. We present here, DIA-MS2pep, a library-free framework for comprehensive peptide identification from DIA data. DIA-MS2pep uses a data-driven algorithm for MS/MS spectrum demultiplexing using the fragments data without the need of a precursor. With a large precursor mass tolerance database search, DIA-MS2pep can identify the peptides and their modified forms. We demonstrate the performance of DIA-MS2pep by comparing it to conventional library-free tools in accuracy and sensitivity of peptide identifications using publicly available DIA datasets of varying samples, including HeLa cell lysates, phosphopeptides, plasma, etc. Compared with data-dependent acquisition-based spectral libraries, spectral libraries built directly from DIA data with DIA-MS2pep improve the accuracy and reproducibility of the quantitative proteome.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
