Jorrit Tjeertes, Carlos A Bacino, Terry Jo Bichell, Lynne M Bird, Mariana Bustamante, Rebecca Crean, Shafali Jeste, Robert W Komorowski, Michelle L Krishnan, Meghan T Miller, David Nobbs, Cesar Ochoa-Lubinoff, Kimberly A Parkerson, Alexander Rotenberg, Anjali Sadhwani, Mark D Shen, Lisa Squassante, Wen-Hann Tan, Brenda Vincenzi, Anne C Wheeler, Joerg F Hipp, Elizabeth Berry-Kravis
{"title":"为安杰曼综合征患者的干预性临床试验制定终点:前瞻性、纵向、观察性临床研究 (FREESIAS)。","authors":"Jorrit Tjeertes, Carlos A Bacino, Terry Jo Bichell, Lynne M Bird, Mariana Bustamante, Rebecca Crean, Shafali Jeste, Robert W Komorowski, Michelle L Krishnan, Meghan T Miller, David Nobbs, Cesar Ochoa-Lubinoff, Kimberly A Parkerson, Alexander Rotenberg, Anjali Sadhwani, Mark D Shen, Lisa Squassante, Wen-Hann Tan, Brenda Vincenzi, Anne C Wheeler, Joerg F Hipp, Elizabeth Berry-Kravis","doi":"10.1186/s11689-023-09494-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by the absence of a functional UBE3A gene, which causes developmental, behavioral, and medical challenges. While currently untreatable, comprehensive data could help identify appropriate endpoints assessing meaningful improvements in clinical trials. Herein are reported the results from the FREESIAS study assessing the feasibility and utility of in-clinic and at-home measures of key AS symptoms.</p><p><strong>Methods: </strong>Fifty-five individuals with AS (aged < 5 years: n = 16, 5-12 years: n = 27, ≥ 18 years: n = 12; deletion genotype: n = 40, nondeletion genotype: n = 15) and 20 typically developing children (aged 1-12 years) were enrolled across six USA sites. Several clinical outcome assessments and digital health technologies were tested, together with overnight 19-lead electroencephalography (EEG) and additional polysomnography (PSG) sensors. Participants were assessed at baseline (Clinic Visit 1), 12 months later (Clinic Visit 2), and during intermittent home visits.</p><p><strong>Results: </strong>The participants achieved high completion rates for the clinical outcome assessments (adherence: 89-100% [Clinic Visit 1]; 76-91% [Clinic Visit 2]) and varied feasibility of and adherence to digital health technologies. The coronavirus disease 2019 (COVID-19) pandemic impacted participants' uptake of and/or adherence to some measures. It also potentially impacted the at-home PSG/EEG recordings, which were otherwise feasible. Participants achieved Bayley-III results comparable to the available natural history data, showing similar scores between individuals aged ≥ 18 and 5-12 years. Also, participants without a deletion generally scored higher on most clinical outcome assessments than participants with a deletion. Furthermore, the observed AS EEG phenotype of excess delta-band power was consistent with prior reports.</p><p><strong>Conclusions: </strong>Although feasible clinical outcome assessments and digital health technologies are reported herein, further improved assessments of meaningful AS change are needed. Despite the COVID-19 pandemic, remote assessments facilitated high adherence levels and the results suggested that at-home PSG/EEG might be a feasible alternative to the in-clinic EEG assessments. Taken altogether, the combination of in-clinic/at-home clinical outcome assessments, digital health technologies, and PSG/EEG may improve protocol adherence, reduce patient burden, and optimize study outcomes in AS and other rare disease populations.</p>","PeriodicalId":16530,"journal":{"name":"Journal of Neurodevelopmental Disorders","volume":"15 1","pages":"22"},"PeriodicalIF":4.0000,"publicationDate":"2023-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10373389/pdf/","citationCount":"0","resultStr":"{\"title\":\"Enabling endpoint development for interventional clinical trials in individuals with Angelman syndrome: a prospective, longitudinal, observational clinical study (FREESIAS).\",\"authors\":\"Jorrit Tjeertes, Carlos A Bacino, Terry Jo Bichell, Lynne M Bird, Mariana Bustamante, Rebecca Crean, Shafali Jeste, Robert W Komorowski, Michelle L Krishnan, Meghan T Miller, David Nobbs, Cesar Ochoa-Lubinoff, Kimberly A Parkerson, Alexander Rotenberg, Anjali Sadhwani, Mark D Shen, Lisa Squassante, Wen-Hann Tan, Brenda Vincenzi, Anne C Wheeler, Joerg F Hipp, Elizabeth Berry-Kravis\",\"doi\":\"10.1186/s11689-023-09494-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by the absence of a functional UBE3A gene, which causes developmental, behavioral, and medical challenges. While currently untreatable, comprehensive data could help identify appropriate endpoints assessing meaningful improvements in clinical trials. Herein are reported the results from the FREESIAS study assessing the feasibility and utility of in-clinic and at-home measures of key AS symptoms.</p><p><strong>Methods: </strong>Fifty-five individuals with AS (aged < 5 years: n = 16, 5-12 years: n = 27, ≥ 18 years: n = 12; deletion genotype: n = 40, nondeletion genotype: n = 15) and 20 typically developing children (aged 1-12 years) were enrolled across six USA sites. Several clinical outcome assessments and digital health technologies were tested, together with overnight 19-lead electroencephalography (EEG) and additional polysomnography (PSG) sensors. Participants were assessed at baseline (Clinic Visit 1), 12 months later (Clinic Visit 2), and during intermittent home visits.</p><p><strong>Results: </strong>The participants achieved high completion rates for the clinical outcome assessments (adherence: 89-100% [Clinic Visit 1]; 76-91% [Clinic Visit 2]) and varied feasibility of and adherence to digital health technologies. The coronavirus disease 2019 (COVID-19) pandemic impacted participants' uptake of and/or adherence to some measures. It also potentially impacted the at-home PSG/EEG recordings, which were otherwise feasible. Participants achieved Bayley-III results comparable to the available natural history data, showing similar scores between individuals aged ≥ 18 and 5-12 years. Also, participants without a deletion generally scored higher on most clinical outcome assessments than participants with a deletion. Furthermore, the observed AS EEG phenotype of excess delta-band power was consistent with prior reports.</p><p><strong>Conclusions: </strong>Although feasible clinical outcome assessments and digital health technologies are reported herein, further improved assessments of meaningful AS change are needed. Despite the COVID-19 pandemic, remote assessments facilitated high adherence levels and the results suggested that at-home PSG/EEG might be a feasible alternative to the in-clinic EEG assessments. Taken altogether, the combination of in-clinic/at-home clinical outcome assessments, digital health technologies, and PSG/EEG may improve protocol adherence, reduce patient burden, and optimize study outcomes in AS and other rare disease populations.</p>\",\"PeriodicalId\":16530,\"journal\":{\"name\":\"Journal of Neurodevelopmental Disorders\",\"volume\":\"15 1\",\"pages\":\"22\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2023-07-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10373389/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neurodevelopmental Disorders\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s11689-023-09494-w\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurodevelopmental Disorders","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s11689-023-09494-w","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Enabling endpoint development for interventional clinical trials in individuals with Angelman syndrome: a prospective, longitudinal, observational clinical study (FREESIAS).
Background: Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by the absence of a functional UBE3A gene, which causes developmental, behavioral, and medical challenges. While currently untreatable, comprehensive data could help identify appropriate endpoints assessing meaningful improvements in clinical trials. Herein are reported the results from the FREESIAS study assessing the feasibility and utility of in-clinic and at-home measures of key AS symptoms.
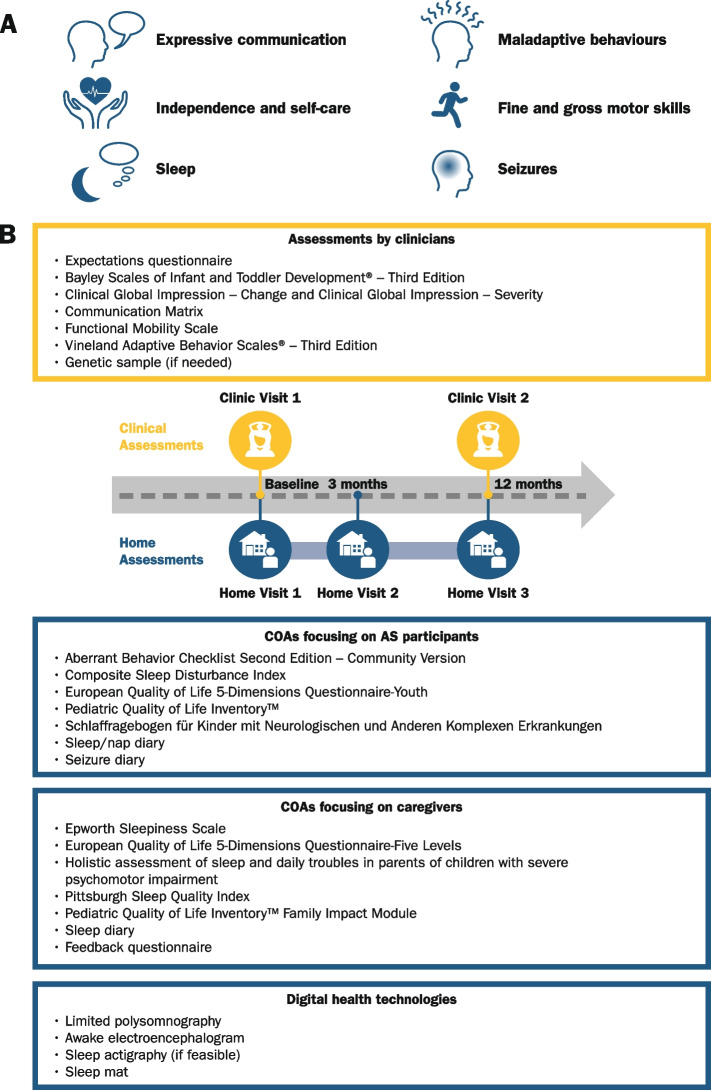
Methods: Fifty-five individuals with AS (aged < 5 years: n = 16, 5-12 years: n = 27, ≥ 18 years: n = 12; deletion genotype: n = 40, nondeletion genotype: n = 15) and 20 typically developing children (aged 1-12 years) were enrolled across six USA sites. Several clinical outcome assessments and digital health technologies were tested, together with overnight 19-lead electroencephalography (EEG) and additional polysomnography (PSG) sensors. Participants were assessed at baseline (Clinic Visit 1), 12 months later (Clinic Visit 2), and during intermittent home visits.
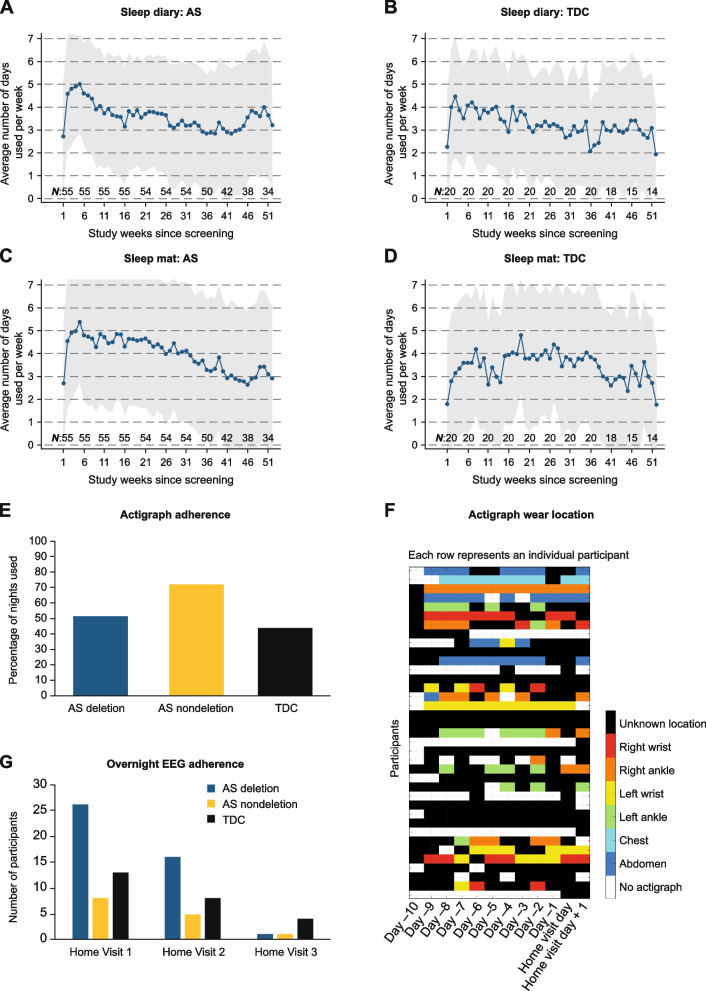
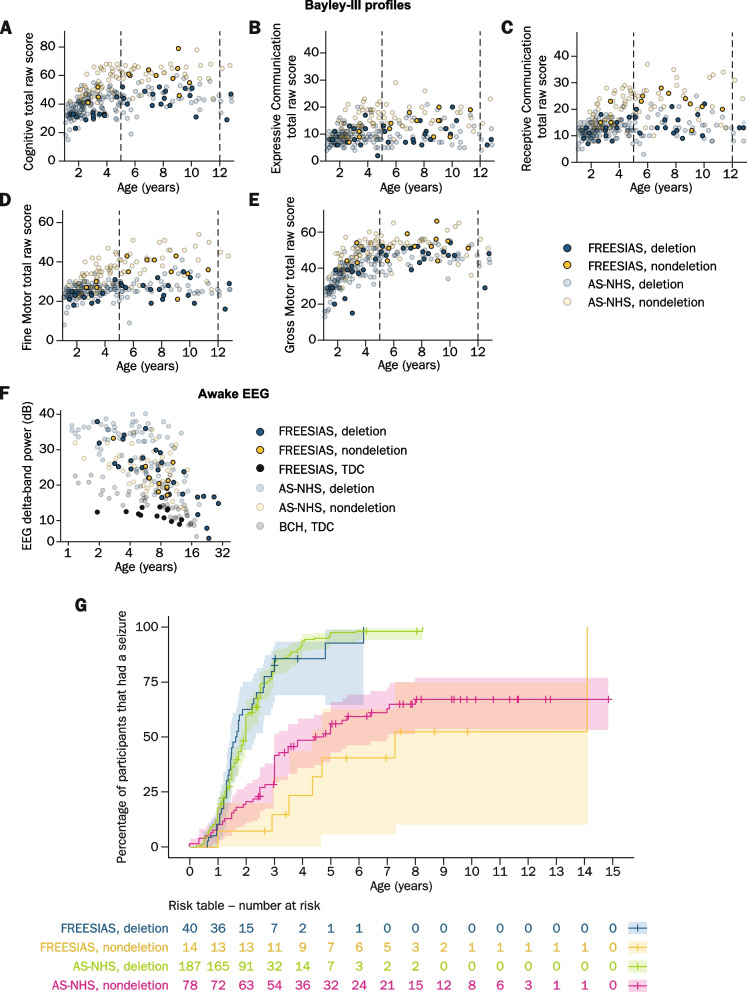
Results: The participants achieved high completion rates for the clinical outcome assessments (adherence: 89-100% [Clinic Visit 1]; 76-91% [Clinic Visit 2]) and varied feasibility of and adherence to digital health technologies. The coronavirus disease 2019 (COVID-19) pandemic impacted participants' uptake of and/or adherence to some measures. It also potentially impacted the at-home PSG/EEG recordings, which were otherwise feasible. Participants achieved Bayley-III results comparable to the available natural history data, showing similar scores between individuals aged ≥ 18 and 5-12 years. Also, participants without a deletion generally scored higher on most clinical outcome assessments than participants with a deletion. Furthermore, the observed AS EEG phenotype of excess delta-band power was consistent with prior reports.
Conclusions: Although feasible clinical outcome assessments and digital health technologies are reported herein, further improved assessments of meaningful AS change are needed. Despite the COVID-19 pandemic, remote assessments facilitated high adherence levels and the results suggested that at-home PSG/EEG might be a feasible alternative to the in-clinic EEG assessments. Taken altogether, the combination of in-clinic/at-home clinical outcome assessments, digital health technologies, and PSG/EEG may improve protocol adherence, reduce patient burden, and optimize study outcomes in AS and other rare disease populations.
期刊介绍:
Journal of Neurodevelopmental Disorders is an open access journal that integrates current, cutting-edge research across a number of disciplines, including neurobiology, genetics, cognitive neuroscience, psychiatry and psychology. The journal’s primary focus is on the pathogenesis of neurodevelopmental disorders including autism, fragile X syndrome, tuberous sclerosis, Turner Syndrome, 22q Deletion Syndrome, Prader-Willi and Angelman Syndrome, Williams syndrome, lysosomal storage diseases, dyslexia, specific language impairment and fetal alcohol syndrome. With the discovery of specific genes underlying neurodevelopmental syndromes, the emergence of powerful tools for studying neural circuitry, and the development of new approaches for exploring molecular mechanisms, interdisciplinary research on the pathogenesis of neurodevelopmental disorders is now increasingly common. Journal of Neurodevelopmental Disorders provides a unique venue for researchers interested in comparing and contrasting mechanisms and characteristics related to the pathogenesis of the full range of neurodevelopmental disorders, sharpening our understanding of the etiology and relevant phenotypes of each condition.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
