{"title":"继发性鼻腔平滑肌肉瘤患者可能患有遗传性视网膜母细胞瘤,伴有RB1和DMXL1的种系相互易位和体细胞TP53突变:1例报告。","authors":"Toshinari Yagi, Harumi Nakamura, Yoji Kukita, Toru Wakamatsu, Hironari Tamiya, Shou Nakai, Makiyo Watanabe, Shigeki Kakunaga, Haruna Takami, Rie Suzuki, Satoshi Takenaka, Yoshiko Hashii","doi":"10.3892/mco.2023.2661","DOIUrl":null,"url":null,"abstract":"<p><p>Retinoblastoma is a common primary intraocular malignant tumor that affects infants and young children. Radiation therapy for hereditary retinoblastoma increases the risk of secondary malignancy. The present report discusses the case of a retinoblastoma survivor who developed secondary leiomyosarcoma 42 years after receiving radiation therapy. The retinoblastoma of the patient was unilateral, and the patient had no family history of the disease. RNA and DNA panel sequencing of the leiomyosarcoma tissue was performed to elucidate the molecular mechanism of this secondary malignancy. The RNA panel sequencing detected a germline reciprocal translocation of RB1 and DMXL1, leading to a diagnosis of possible hereditary retinoblastoma. Furthermore, it detected a somatic fusion gene (RAD51-KNL1). The DNA panel sequencing identified various germline or somatic variants, including a somatic splice acceptor site mutation of TP53. We hypothesized that the molecular mechanism of the secondary malignancy of this patient was the combination of a germline reciprocal translocation of RB1 and DMXL1 and the accumulation of various somatic mutations containing the splice acceptor site mutation of TP53, which ultimately led to the development of a secondary leiomyosarcoma. Further prospective investigations are necessary to fully understand the role of reciprocal translocation of RB1 and DMXL1 or other mutations in the tumorigenesis of second malignancies in patients with hereditary retinoblastoma.</p>","PeriodicalId":18737,"journal":{"name":"Molecular and clinical oncology","volume":"19 2","pages":"65"},"PeriodicalIF":1.4000,"publicationDate":"2023-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0d/0d/mco-19-02-02661.PMC10407467.pdf","citationCount":"0","resultStr":"{\"title\":\"Secondary leiomyosarcoma of the nasal cavity in a treated patient with possible hereditary retinoblastoma with germline reciprocal translocation of RB1 and DMXL1 and somatic TP53 mutation: A case report.\",\"authors\":\"Toshinari Yagi, Harumi Nakamura, Yoji Kukita, Toru Wakamatsu, Hironari Tamiya, Shou Nakai, Makiyo Watanabe, Shigeki Kakunaga, Haruna Takami, Rie Suzuki, Satoshi Takenaka, Yoshiko Hashii\",\"doi\":\"10.3892/mco.2023.2661\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Retinoblastoma is a common primary intraocular malignant tumor that affects infants and young children. Radiation therapy for hereditary retinoblastoma increases the risk of secondary malignancy. The present report discusses the case of a retinoblastoma survivor who developed secondary leiomyosarcoma 42 years after receiving radiation therapy. The retinoblastoma of the patient was unilateral, and the patient had no family history of the disease. RNA and DNA panel sequencing of the leiomyosarcoma tissue was performed to elucidate the molecular mechanism of this secondary malignancy. The RNA panel sequencing detected a germline reciprocal translocation of RB1 and DMXL1, leading to a diagnosis of possible hereditary retinoblastoma. Furthermore, it detected a somatic fusion gene (RAD51-KNL1). The DNA panel sequencing identified various germline or somatic variants, including a somatic splice acceptor site mutation of TP53. We hypothesized that the molecular mechanism of the secondary malignancy of this patient was the combination of a germline reciprocal translocation of RB1 and DMXL1 and the accumulation of various somatic mutations containing the splice acceptor site mutation of TP53, which ultimately led to the development of a secondary leiomyosarcoma. Further prospective investigations are necessary to fully understand the role of reciprocal translocation of RB1 and DMXL1 or other mutations in the tumorigenesis of second malignancies in patients with hereditary retinoblastoma.</p>\",\"PeriodicalId\":18737,\"journal\":{\"name\":\"Molecular and clinical oncology\",\"volume\":\"19 2\",\"pages\":\"65\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2023-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/0d/0d/mco-19-02-02661.PMC10407467.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular and clinical oncology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3892/mco.2023.2661\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and clinical oncology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3892/mco.2023.2661","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"ONCOLOGY","Score":null,"Total":0}
Secondary leiomyosarcoma of the nasal cavity in a treated patient with possible hereditary retinoblastoma with germline reciprocal translocation of RB1 and DMXL1 and somatic TP53 mutation: A case report.
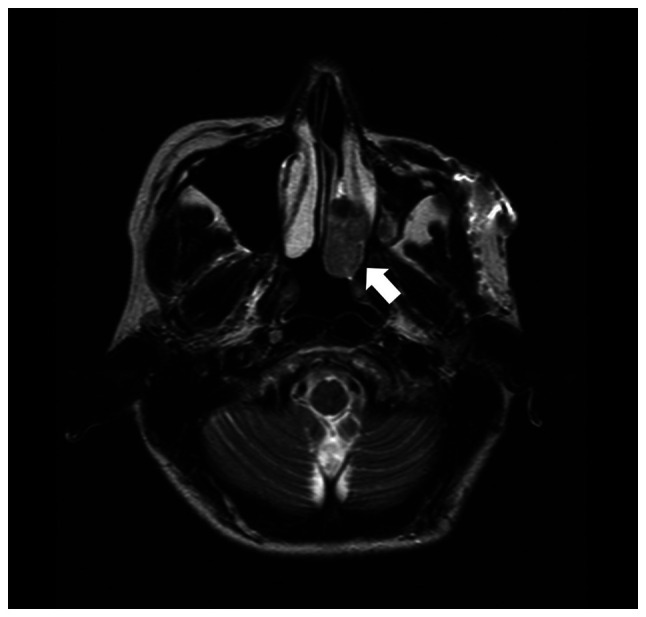
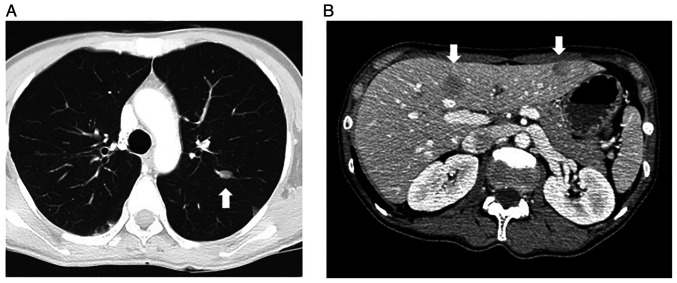
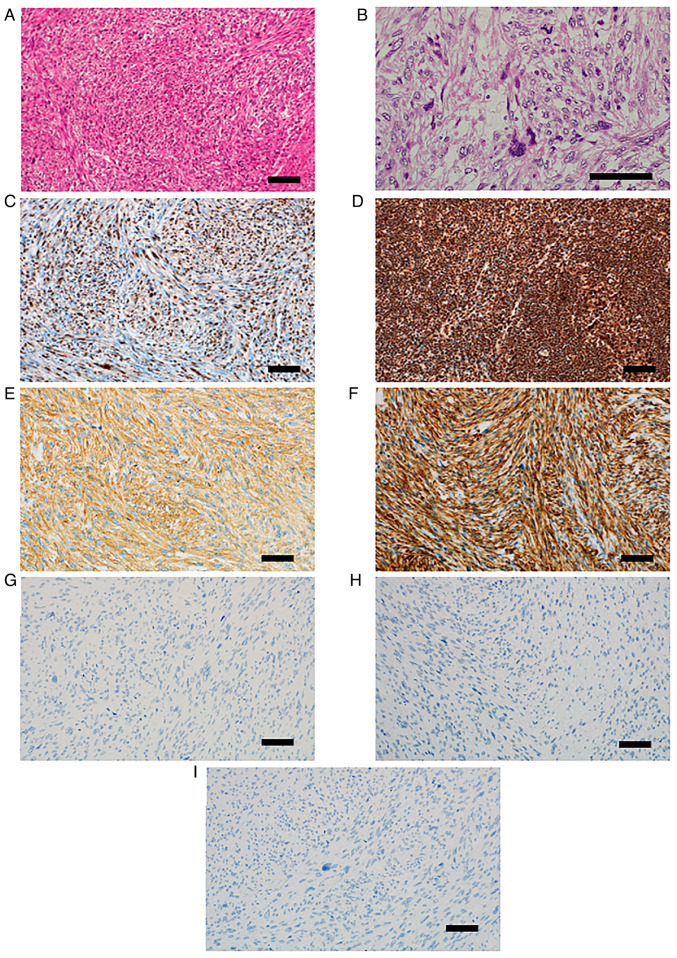
Retinoblastoma is a common primary intraocular malignant tumor that affects infants and young children. Radiation therapy for hereditary retinoblastoma increases the risk of secondary malignancy. The present report discusses the case of a retinoblastoma survivor who developed secondary leiomyosarcoma 42 years after receiving radiation therapy. The retinoblastoma of the patient was unilateral, and the patient had no family history of the disease. RNA and DNA panel sequencing of the leiomyosarcoma tissue was performed to elucidate the molecular mechanism of this secondary malignancy. The RNA panel sequencing detected a germline reciprocal translocation of RB1 and DMXL1, leading to a diagnosis of possible hereditary retinoblastoma. Furthermore, it detected a somatic fusion gene (RAD51-KNL1). The DNA panel sequencing identified various germline or somatic variants, including a somatic splice acceptor site mutation of TP53. We hypothesized that the molecular mechanism of the secondary malignancy of this patient was the combination of a germline reciprocal translocation of RB1 and DMXL1 and the accumulation of various somatic mutations containing the splice acceptor site mutation of TP53, which ultimately led to the development of a secondary leiomyosarcoma. Further prospective investigations are necessary to fully understand the role of reciprocal translocation of RB1 and DMXL1 or other mutations in the tumorigenesis of second malignancies in patients with hereditary retinoblastoma.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
