Dmitry Zankov, Timur Madzhidov, Igor Baskin, Alexandre Varnek
{"title":"Conjugated quantitative structure-property relationship models: Prediction of kinetic characteristics linked by the Arrhenius equation.","authors":"Dmitry Zankov, Timur Madzhidov, Igor Baskin, Alexandre Varnek","doi":"10.1002/minf.202200275","DOIUrl":null,"url":null,"abstract":"<p><p>Conjugated QSPR models for reactions integrate fundamental chemical laws expressed by mathematical equations with machine learning algorithms. Herein we present a methodology for building conjugated QSPR models integrated with the Arrhenius equation. Conjugated QSPR models were used to predict kinetic characteristics of cycloaddition reactions related by the Arrhenius equation: rate constant <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>k</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}k}$</annotation> </semantics> </math> , pre-exponential factor <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>A</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}A}$</annotation> </semantics> </math> , and activation energy <math> <semantics><msub><mi>E</mi> <mi>a</mi></msub> <annotation>${{E}_{{\\rm a}}}$</annotation> </semantics> </math> . They were benchmarked against single-task (individual and equation-based models) and multi-task models. In individual models, all characteristics were modeled separately, while in multi-task models <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>k</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}k}$</annotation> </semantics> </math> , <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>A</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}A}$</annotation> </semantics> </math> and <math> <semantics><msub><mi>E</mi> <mi>a</mi></msub> <annotation>${{E}_{{\\rm a}}}$</annotation> </semantics> </math> were treated cooperatively. An equation-based model assessed <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>k</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}k}$</annotation> </semantics> </math> using the Arrhenius equation and <math> <semantics><mrow><mi>l</mi> <mi>o</mi> <mi>g</mi> <mi>A</mi></mrow> <annotation>${{\\rm l}{\\rm o}{\\rm g}A}$</annotation> </semantics> </math> and <math> <semantics><msub><mi>E</mi> <mi>a</mi></msub> <annotation>${{E}_{{\\rm a}}}$</annotation> </semantics> </math> values predicted by individual models. It has been demonstrated that the conjugated QSPR models can accurately predict the reaction rate constants at extreme temperatures, at which reaction rate constants hardly can be measured experimentally. Also, in the case of small training sets conjugated models are more robust than related single-task approaches.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":" ","pages":"e2200275"},"PeriodicalIF":3.1000,"publicationDate":"2023-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202200275","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/8/21 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
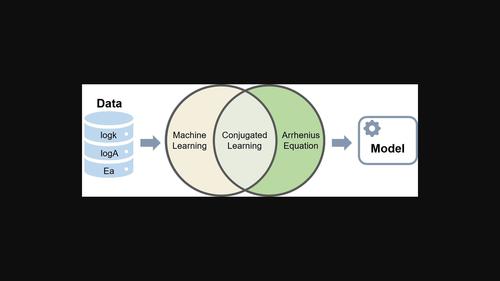
Conjugated QSPR models for reactions integrate fundamental chemical laws expressed by mathematical equations with machine learning algorithms. Herein we present a methodology for building conjugated QSPR models integrated with the Arrhenius equation. Conjugated QSPR models were used to predict kinetic characteristics of cycloaddition reactions related by the Arrhenius equation: rate constant , pre-exponential factor , and activation energy . They were benchmarked against single-task (individual and equation-based models) and multi-task models. In individual models, all characteristics were modeled separately, while in multi-task models , and were treated cooperatively. An equation-based model assessed using the Arrhenius equation and and values predicted by individual models. It has been demonstrated that the conjugated QSPR models can accurately predict the reaction rate constants at extreme temperatures, at which reaction rate constants hardly can be measured experimentally. Also, in the case of small training sets conjugated models are more robust than related single-task approaches.
反应的共轭QSPR模型将数学方程表达的基本化学定律与机器学习算法相结合。在此,我们提出了一种结合阿伦尼斯方程建立共轭QSPR模型的方法。共轭QSPR模型用于预测与Arrhenius方程相关的环加成反应的动力学特性:速率常数l o g k${\rm l}{\rmo}{{\RMg}k}$、指数前因子l o g A${\ rml}和活化能E A${{E}_{{\rm a}}$。它们以单任务(基于个体和方程的模型)和多任务模型为基准。在单独的模型中,所有特征都是单独建模的,而在多任务模型中,l o g k${\rm l}${{E}_{\rma}}$得到了合作处理。一个基于方程的模型使用Arrhenius方程和l o g A${\rm l}{\rm o}A}$和E${{E}_{{\rma}}}}$由各个模型预测的值。研究表明,共轭QSPR模型可以准确预测极端温度下的反应速率常数,而在极端温度下几乎无法通过实验测量反应速率常数。此外,在小训练集的情况下,共轭模型比相关的单任务方法更稳健。
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
