{"title":"gmXtal: Cooking Crystals with GROMACS","authors":"Pavel Buslaev, Gerrit Groenhof","doi":"10.1007/s10930-023-10141-5","DOIUrl":null,"url":null,"abstract":"<div><p>Molecular dynamics (MD) simulations are routinely performed of biomolecules in solution, because this is their native environment. However, the structures used in such simulations are often obtained with X-ray crystallography, which provides the atomic coordinates of the biomolecule in a crystal environment. With the advent of free electron lasers and time-resolved techniques, X-ray crystallography can now also access metastable states that are intermediates in a biochemical process. Such experiments provide additional data, which can be used, for example, to optimize MD force fields. Doing so requires that the simulation of the biomolecule is also performed in the crystal environment. However, in contrast to simulations of biomolecules in solution, setting up a crystal is challenging. In particular, because not all solvent molecules are resolved in X-ray crystallography, adding a suitable number of solvent molecules, such that the properties of the crystallographic unit cell are preserved in the simulation, can be difficult and typically is a trial-and-error based procedure requiring manual interventions. Such interventions preclude high throughput applications. To overcome this bottleneck, we introduce <b>gmXtal</b>, a tool for setting up crystal simulations for MD simulations with GROMACS. With the information from the protein data bank (rcsb.org) <b>gmXtal</b> automatically (i) builds the crystallographic unit cell; (ii) sets the protonation of titratable residues; (iii) builds missing residues that were not resolved experimentally; and (iv) adds an appropriate number of solvent molecules to the system. <b>gmXtal</b> is available as a standalone tool https://gitlab.com/pbuslaev/gmxtal.</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":793,"journal":{"name":"The Protein Journal","volume":"43 2","pages":"200 - 206"},"PeriodicalIF":1.4000,"publicationDate":"2023-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11058868/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Protein Journal","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1007/s10930-023-10141-5","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
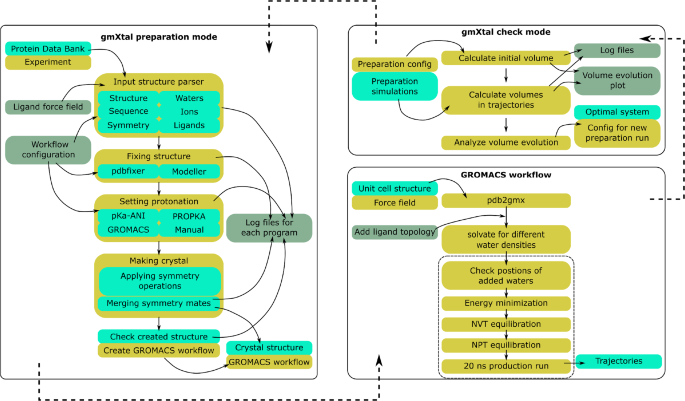
Molecular dynamics (MD) simulations are routinely performed of biomolecules in solution, because this is their native environment. However, the structures used in such simulations are often obtained with X-ray crystallography, which provides the atomic coordinates of the biomolecule in a crystal environment. With the advent of free electron lasers and time-resolved techniques, X-ray crystallography can now also access metastable states that are intermediates in a biochemical process. Such experiments provide additional data, which can be used, for example, to optimize MD force fields. Doing so requires that the simulation of the biomolecule is also performed in the crystal environment. However, in contrast to simulations of biomolecules in solution, setting up a crystal is challenging. In particular, because not all solvent molecules are resolved in X-ray crystallography, adding a suitable number of solvent molecules, such that the properties of the crystallographic unit cell are preserved in the simulation, can be difficult and typically is a trial-and-error based procedure requiring manual interventions. Such interventions preclude high throughput applications. To overcome this bottleneck, we introduce gmXtal, a tool for setting up crystal simulations for MD simulations with GROMACS. With the information from the protein data bank (rcsb.org) gmXtal automatically (i) builds the crystallographic unit cell; (ii) sets the protonation of titratable residues; (iii) builds missing residues that were not resolved experimentally; and (iv) adds an appropriate number of solvent molecules to the system. gmXtal is available as a standalone tool https://gitlab.com/pbuslaev/gmxtal.
期刊介绍:
The Protein Journal (formerly the Journal of Protein Chemistry) publishes original research work on all aspects of proteins and peptides. These include studies concerned with covalent or three-dimensional structure determination (X-ray, NMR, cryoEM, EPR/ESR, optical methods, etc.), computational aspects of protein structure and function, protein folding and misfolding, assembly, genetics, evolution, proteomics, molecular biology, protein engineering, protein nanotechnology, protein purification and analysis and peptide synthesis, as well as the elucidation and interpretation of the molecular bases of biological activities of proteins and peptides. We accept original research papers, reviews, mini-reviews, hypotheses, opinion papers, and letters to the editor.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
