Anna Zhukova, Frédéric Hecht, Yvon Maday, Olivier Gascuel
{"title":"Fast and Accurate Maximum-Likelihood Estimation of Multi-Type Birth-Death Epidemiological Models from Phylogenetic Trees.","authors":"Anna Zhukova, Frédéric Hecht, Yvon Maday, Olivier Gascuel","doi":"10.1093/sysbio/syad059","DOIUrl":null,"url":null,"abstract":"<p><p>Multi-type birth-death (MTBD) models are phylodynamic analogies of compartmental models in classical epidemiology. They serve to infer such epidemiological parameters as the average number of secondary infections Re and the infectious time from a phylogenetic tree (a genealogy of pathogen sequences). The representatives of this model family focus on various aspects of pathogen epidemics. For instance, the birth-death exposed-infectious (BDEI) model describes the transmission of pathogens featuring an incubation period (when there is a delay between the moment of infection and becoming infectious, as for Ebola and SARS-CoV-2), and permits its estimation along with other parameters. With constantly growing sequencing data, MTBD models should be extremely useful for unravelling information on pathogen epidemics. However, existing implementations of these models in a phylodynamic framework have not yet caught up with the sequencing speed. Computing time and numerical instability issues limit their applicability to medium data sets (≤ 500 samples), while the accuracy of estimations should increase with more data. We propose a new highly parallelizable formulation of ordinary differential equations for MTBD models. We also extend them to forests to represent situations when a (sub-)epidemic started from several cases (e.g., multiple introductions to a country). We implemented it for the BDEI model in a maximum likelihood framework using a combination of numerical analysis methods for efficient equation resolution. Our implementation estimates epidemiological parameter values and their confidence intervals in two minutes on a phylogenetic tree of 10,000 samples. Comparison to the existing implementations on simulated data shows that it is not only much faster but also more accurate. An application of our tool to the 2014 Ebola epidemic in Sierra-Leone is also convincing, with very fast calculation and precise estimates. As MTBD models are closely related to Cladogenetic State Speciation and Extinction (ClaSSE)-like models, our findings could also be easily transferred to the macroevolution domain.</p>","PeriodicalId":22120,"journal":{"name":"Systematic Biology","volume":" ","pages":"1387-1402"},"PeriodicalIF":5.7000,"publicationDate":"2023-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10924745/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Systematic Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/sysbio/syad059","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
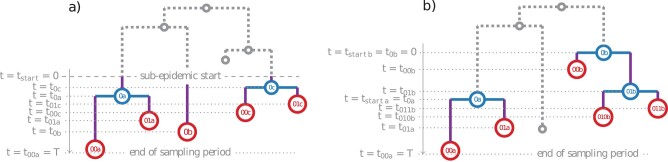
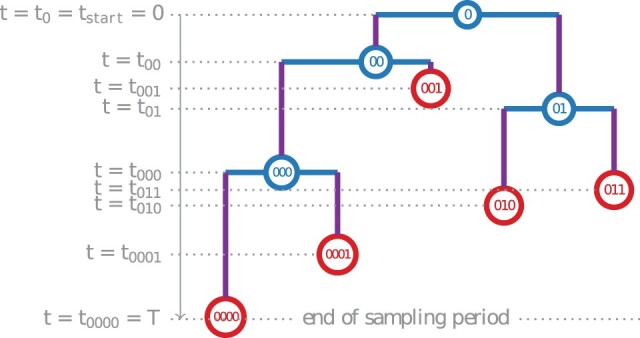
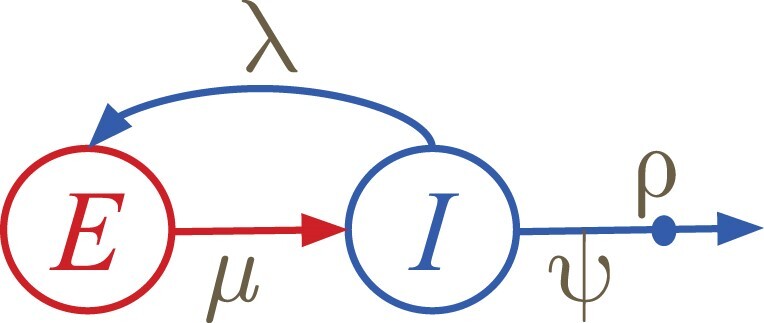
Multi-type birth-death (MTBD) models are phylodynamic analogies of compartmental models in classical epidemiology. They serve to infer such epidemiological parameters as the average number of secondary infections Re and the infectious time from a phylogenetic tree (a genealogy of pathogen sequences). The representatives of this model family focus on various aspects of pathogen epidemics. For instance, the birth-death exposed-infectious (BDEI) model describes the transmission of pathogens featuring an incubation period (when there is a delay between the moment of infection and becoming infectious, as for Ebola and SARS-CoV-2), and permits its estimation along with other parameters. With constantly growing sequencing data, MTBD models should be extremely useful for unravelling information on pathogen epidemics. However, existing implementations of these models in a phylodynamic framework have not yet caught up with the sequencing speed. Computing time and numerical instability issues limit their applicability to medium data sets (≤ 500 samples), while the accuracy of estimations should increase with more data. We propose a new highly parallelizable formulation of ordinary differential equations for MTBD models. We also extend them to forests to represent situations when a (sub-)epidemic started from several cases (e.g., multiple introductions to a country). We implemented it for the BDEI model in a maximum likelihood framework using a combination of numerical analysis methods for efficient equation resolution. Our implementation estimates epidemiological parameter values and their confidence intervals in two minutes on a phylogenetic tree of 10,000 samples. Comparison to the existing implementations on simulated data shows that it is not only much faster but also more accurate. An application of our tool to the 2014 Ebola epidemic in Sierra-Leone is also convincing, with very fast calculation and precise estimates. As MTBD models are closely related to Cladogenetic State Speciation and Extinction (ClaSSE)-like models, our findings could also be easily transferred to the macroevolution domain.
期刊介绍:
Systematic Biology is the bimonthly journal of the Society of Systematic Biologists. Papers for the journal are original contributions to the theory, principles, and methods of systematics as well as phylogeny, evolution, morphology, biogeography, paleontology, genetics, and the classification of all living things. A Points of View section offers a forum for discussion, while book reviews and announcements of general interest are also featured.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
