Soyoung Jeon, Ying Chu Lo, Libby M Morimoto, Catherine Metayer, Xiaomei Ma, Joseph L Wiemels, Adam J de Smith, Charleston W K Chiang
{"title":"Evaluating genomic polygenic risk scores for childhood acute lymphoblastic leukemia in Latinos.","authors":"Soyoung Jeon, Ying Chu Lo, Libby M Morimoto, Catherine Metayer, Xiaomei Ma, Joseph L Wiemels, Adam J de Smith, Charleston W K Chiang","doi":"10.1016/j.xhgg.2023.100239","DOIUrl":null,"url":null,"abstract":"<p><p>The utility of polygenic risk score (PRS) models has not been comprehensively evaluated for childhood acute lymphoblastic leukemia (ALL), the most common type of cancer in children. Previous PRS models for ALL were based on significant loci observed in genome-wide association studies (GWASs), even though genomic PRS models have been shown to improve prediction performance for a number of complex diseases. In the United States, Latino (LAT) children have the highest risk of ALL, but the transferability of PRS models to LAT children has not been studied. In this study, we constructed and evaluated genomic PRS models based on either non-Latino White (NLW) GWAS or a multi-ancestry GWAS. We found that the best PRS models performed similarly between held-out NLW and LAT samples (PseudoR<sup>2</sup> = 0.086 ± 0.023 in NLW vs. 0.060 ± 0.020 in LAT), and can be improved for LAT if we performed GWAS in LAT-only (PseudoR<sup>2</sup> = 0.116 ± 0.026) or multi-ancestry samples (PseudoR<sup>2</sup> = 0.131 ± 0.025). However, the best genomic models currently do not have better prediction accuracy than a conventional model using all known ALL-associated loci in the literature (PseudoR<sup>2</sup> = 0.166 ± 0.025), which includes loci from GWAS populations that we could not access to train genomic PRS models. Our results suggest that larger and more inclusive GWASs may be needed for genomic PRS to be useful for ALL. Moreover, the comparable performance between populations may suggest a more oligogenic architecture for ALL, where some large effect loci may be shared between populations. Future PRS models that move away from the infinite causal loci assumption may further improve PRS for ALL.</p>","PeriodicalId":34530,"journal":{"name":"HGG Advances","volume":" ","pages":"100239"},"PeriodicalIF":3.6000,"publicationDate":"2023-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/af/e8/main.PMC10550840.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HGG Advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.xhgg.2023.100239","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/14 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
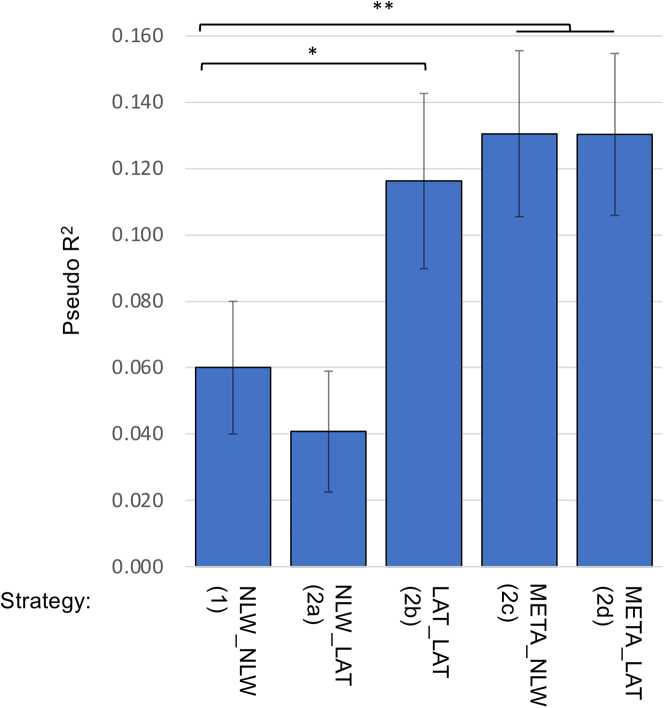
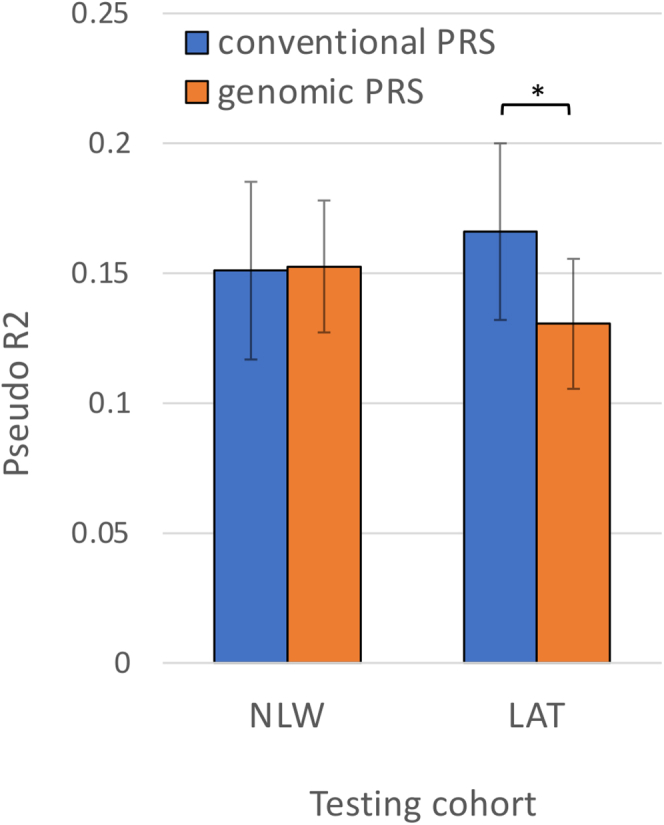
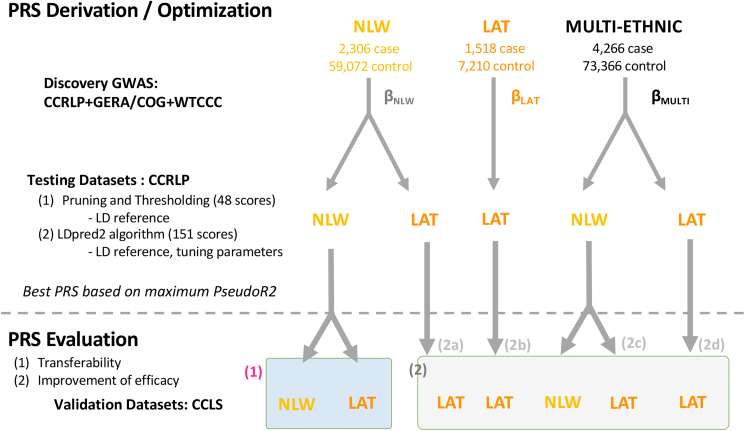
The utility of polygenic risk score (PRS) models has not been comprehensively evaluated for childhood acute lymphoblastic leukemia (ALL), the most common type of cancer in children. Previous PRS models for ALL were based on significant loci observed in genome-wide association studies (GWASs), even though genomic PRS models have been shown to improve prediction performance for a number of complex diseases. In the United States, Latino (LAT) children have the highest risk of ALL, but the transferability of PRS models to LAT children has not been studied. In this study, we constructed and evaluated genomic PRS models based on either non-Latino White (NLW) GWAS or a multi-ancestry GWAS. We found that the best PRS models performed similarly between held-out NLW and LAT samples (PseudoR2 = 0.086 ± 0.023 in NLW vs. 0.060 ± 0.020 in LAT), and can be improved for LAT if we performed GWAS in LAT-only (PseudoR2 = 0.116 ± 0.026) or multi-ancestry samples (PseudoR2 = 0.131 ± 0.025). However, the best genomic models currently do not have better prediction accuracy than a conventional model using all known ALL-associated loci in the literature (PseudoR2 = 0.166 ± 0.025), which includes loci from GWAS populations that we could not access to train genomic PRS models. Our results suggest that larger and more inclusive GWASs may be needed for genomic PRS to be useful for ALL. Moreover, the comparable performance between populations may suggest a more oligogenic architecture for ALL, where some large effect loci may be shared between populations. Future PRS models that move away from the infinite causal loci assumption may further improve PRS for ALL.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
