Yongming Fu, Tuoyu Cao, Xiaorui Zou, Yubing Ye, Youhong Liu, Yuchong Peng, Tanggang Deng, Linglong Yin, Xiong Li
{"title":"AKT1 regulates UHRF1 protein stability and promotes the resistance to abiraterone in prostate cancer.","authors":"Yongming Fu, Tuoyu Cao, Xiaorui Zou, Yubing Ye, Youhong Liu, Yuchong Peng, Tanggang Deng, Linglong Yin, Xiong Li","doi":"10.1038/s41389-022-00446-y","DOIUrl":null,"url":null,"abstract":"<p><p>Oncogenic activation of PI3K/AKT signaling pathway, together with epigenetic aberrations are the characters of castration-resistant prostate cancer (CRPC). UHRF1 as a key epigenetic regulator, plays a critical role in prostate cancer (PCa) development, and its expression is positively correlated with the degree of malignancy. In this present study we investigated the potential regulatory mechanism of AKT1 on UHRF1, and further validated the in vitro and in vivo anticancer efficacy of AKT phosphorylation inhibitor MK2206 in combination with abiraterone. Both UHRF1 and p-AKT aberrantly overexpressed in the abiraterone-resistant PCa cells. Further studies revealed that AKT1 protein interacts with UHRF1, and AKT1 directly phosphorylates UHRF1 via the site Thr-210. MK2206 induced UHRF1 protein degradation by inhibiting AKT1-induced UHRF1 phosphorylation, and then reduced the interaction between UHRF1 and deubiquitinase USP7, while promoted the interaction between UHRF1 and E3 ubiquitin protein ligase BTRC. MK2206 significantly promoted the sensitivity of abiraterone-refractory PCa cells and xenografts to abiraterone by decreasing UHRF1 protein level, and reversed the phenotype of NEPC, evently induced cellular senescence and cell apoptosis. Altogether, our present study for the first time revealed a novel molecular mechanism of abiraterone resistance through PI3K/AKT-UHRF1 pathway, and provided a novel therapeutic modality by targeting PI3K/AKT1 to promote the drug sensitivity of abiraterone in PCa patients.</p>","PeriodicalId":19489,"journal":{"name":"Oncogenesis","volume":"12 1","pages":"1"},"PeriodicalIF":6.4000,"publicationDate":"2023-01-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9807647/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Oncogenesis","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41389-022-00446-y","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 3
Abstract
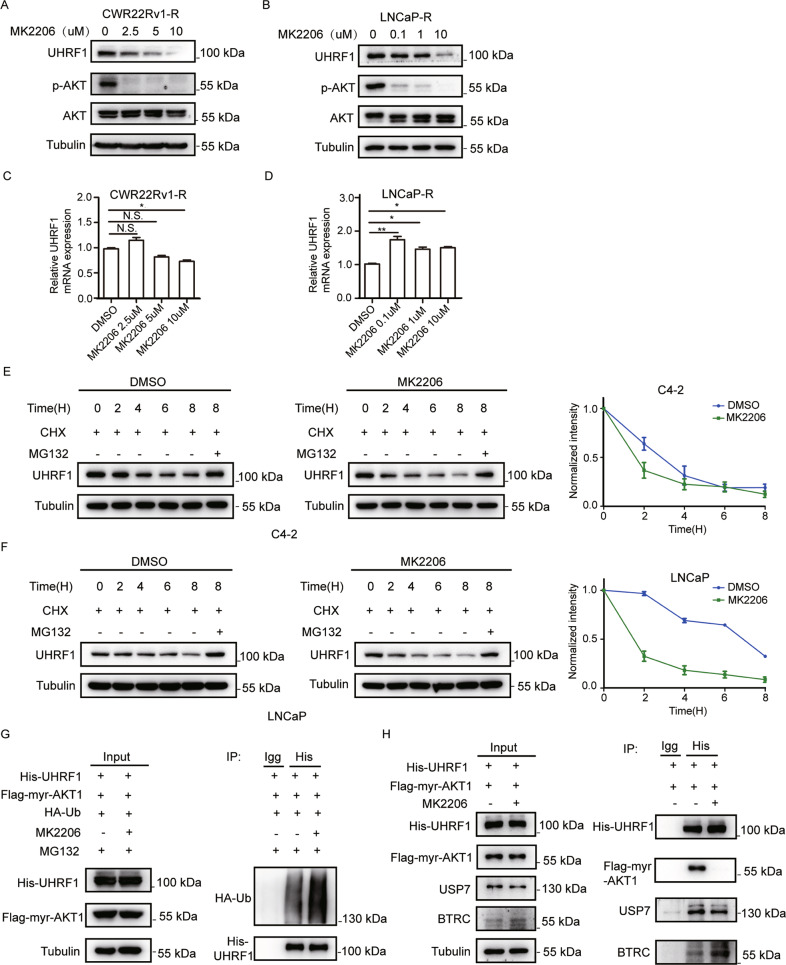
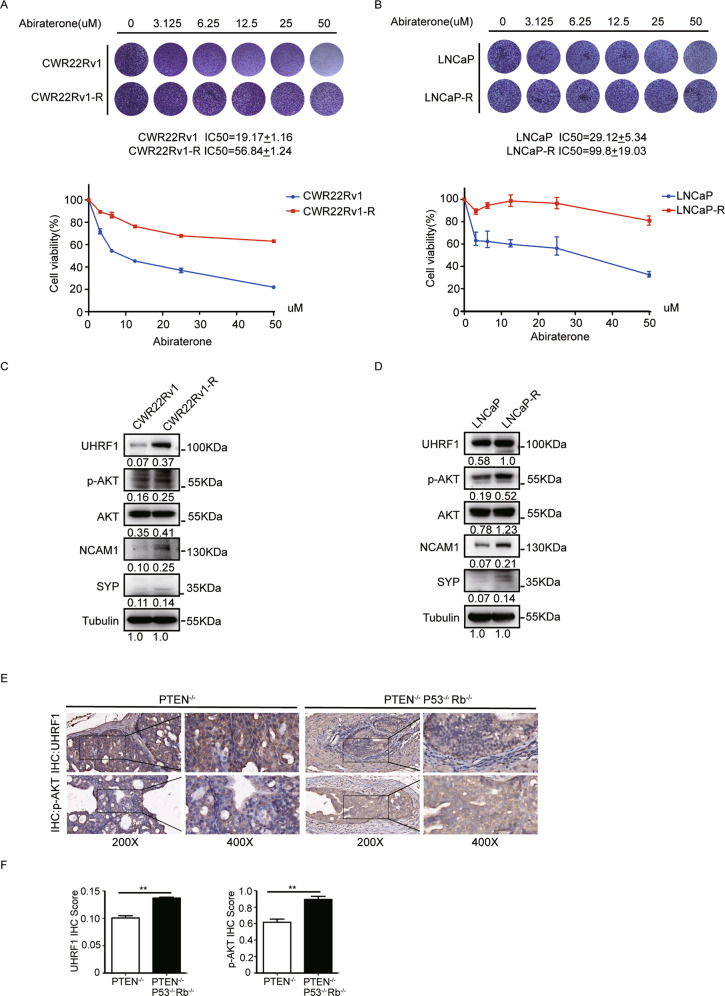
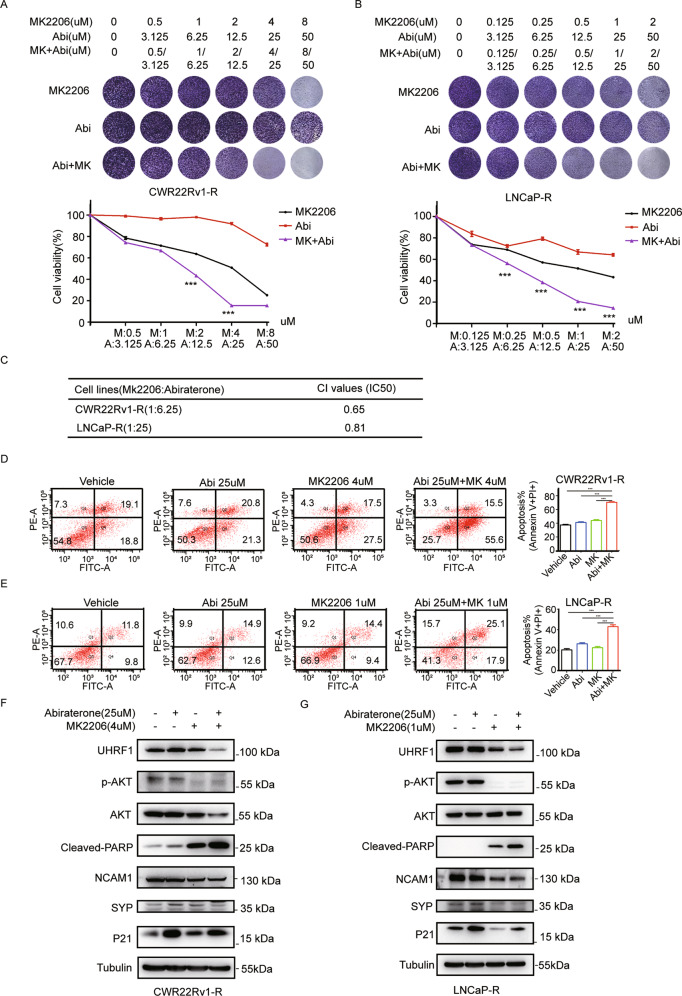
Oncogenic activation of PI3K/AKT signaling pathway, together with epigenetic aberrations are the characters of castration-resistant prostate cancer (CRPC). UHRF1 as a key epigenetic regulator, plays a critical role in prostate cancer (PCa) development, and its expression is positively correlated with the degree of malignancy. In this present study we investigated the potential regulatory mechanism of AKT1 on UHRF1, and further validated the in vitro and in vivo anticancer efficacy of AKT phosphorylation inhibitor MK2206 in combination with abiraterone. Both UHRF1 and p-AKT aberrantly overexpressed in the abiraterone-resistant PCa cells. Further studies revealed that AKT1 protein interacts with UHRF1, and AKT1 directly phosphorylates UHRF1 via the site Thr-210. MK2206 induced UHRF1 protein degradation by inhibiting AKT1-induced UHRF1 phosphorylation, and then reduced the interaction between UHRF1 and deubiquitinase USP7, while promoted the interaction between UHRF1 and E3 ubiquitin protein ligase BTRC. MK2206 significantly promoted the sensitivity of abiraterone-refractory PCa cells and xenografts to abiraterone by decreasing UHRF1 protein level, and reversed the phenotype of NEPC, evently induced cellular senescence and cell apoptosis. Altogether, our present study for the first time revealed a novel molecular mechanism of abiraterone resistance through PI3K/AKT-UHRF1 pathway, and provided a novel therapeutic modality by targeting PI3K/AKT1 to promote the drug sensitivity of abiraterone in PCa patients.
期刊介绍:
Oncogenesis is a peer-reviewed open access online journal that publishes full-length papers, reviews, and short communications exploring the molecular basis of cancer and related phenomena. It seeks to promote diverse and integrated areas of molecular biology, cell biology, oncology, and genetics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
