Roman Tremmel, Yitian Zhou, Matthias Schwab, Volker M Lauschke
{"title":"Structural variation of the coding and non-coding human pharmacogenome.","authors":"Roman Tremmel, Yitian Zhou, Matthias Schwab, Volker M Lauschke","doi":"10.1038/s41525-023-00371-y","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic variants in drug targets and genes encoding factors involved in drug absorption, distribution, metabolism and excretion (ADME) can have pronounced impacts on drug pharmacokinetics, response, and toxicity. While the landscape of genetic variability at the level of single nucleotide variants (SNVs) has been extensively studied in these pharmacogenetic loci, their structural variation is only poorly understood. Thus, we systematically analyzed the genetic structural variability across 908 pharmacogenes (344 ADME genes and 564 drug targets) based on publicly available whole genome sequencing data from 10,847 unrelated individuals. Overall, we extracted 14,984 distinct structural variants (SVs) ranging in size from 50 bp to 106 Mb. Each individual harbored on average 10.3 and 1.5 SVs with putative functional effects that affected the coding regions of ADME genes and drug targets, respectively. In addition, by cross-referencing pharmacogenomic SVs with experimentally determined binding data of 224 transcription factors across 130 cell types, we identified 1276 non-coding SVs that overlapped with gene regulatory elements. Based on these data, we estimate that non-coding structural variants account for 22% of the genetically encoded pharmacogenomic variability. Combined, these analyses provide the first comprehensive map of structural variability across pharmacogenes, derive estimates for the functional impact of non-coding SVs and incentivize the incorporation of structural genomic data into personalized drug response predictions.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"8 1","pages":"24"},"PeriodicalIF":4.7000,"publicationDate":"2023-09-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10491600/pdf/","citationCount":"1","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00371-y","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 1
Abstract
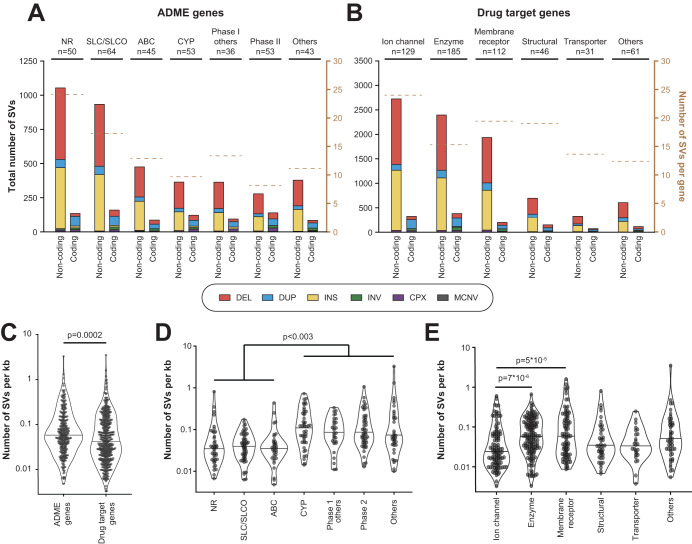
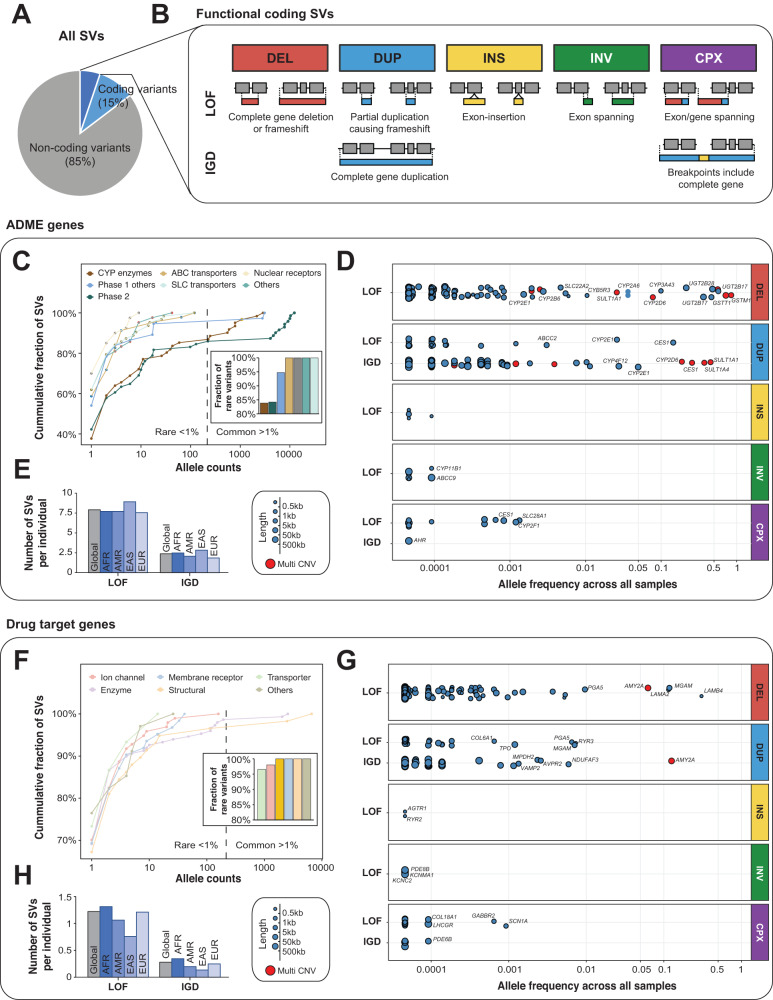
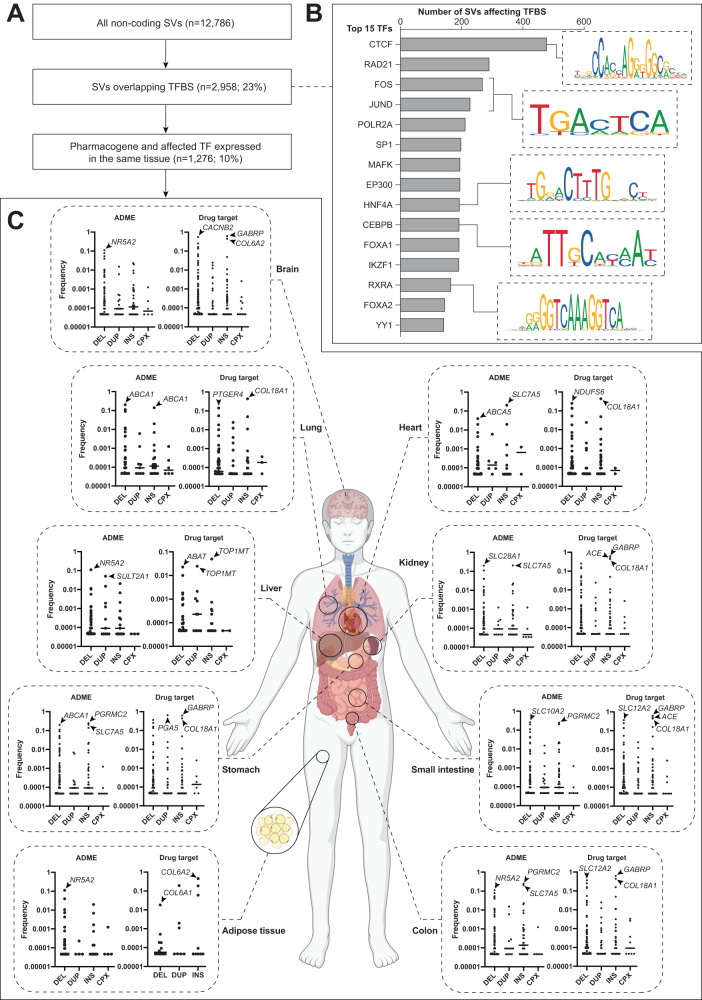
Genetic variants in drug targets and genes encoding factors involved in drug absorption, distribution, metabolism and excretion (ADME) can have pronounced impacts on drug pharmacokinetics, response, and toxicity. While the landscape of genetic variability at the level of single nucleotide variants (SNVs) has been extensively studied in these pharmacogenetic loci, their structural variation is only poorly understood. Thus, we systematically analyzed the genetic structural variability across 908 pharmacogenes (344 ADME genes and 564 drug targets) based on publicly available whole genome sequencing data from 10,847 unrelated individuals. Overall, we extracted 14,984 distinct structural variants (SVs) ranging in size from 50 bp to 106 Mb. Each individual harbored on average 10.3 and 1.5 SVs with putative functional effects that affected the coding regions of ADME genes and drug targets, respectively. In addition, by cross-referencing pharmacogenomic SVs with experimentally determined binding data of 224 transcription factors across 130 cell types, we identified 1276 non-coding SVs that overlapped with gene regulatory elements. Based on these data, we estimate that non-coding structural variants account for 22% of the genetically encoded pharmacogenomic variability. Combined, these analyses provide the first comprehensive map of structural variability across pharmacogenes, derive estimates for the functional impact of non-coding SVs and incentivize the incorporation of structural genomic data into personalized drug response predictions.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
