Thales C Nepomuceno, Tzeh Keong Foo, Marcy E Richardson, John Michael O Ranola, Jamie Weyandt, Matthew J Varga, Amaya Alarcon, Diana Gutierrez, Anna von Wachenfeldt, Daniel Eriksson, Raymond Kim, Susan Armel, Edwin Iversen, Fergus J Couch, Åke Borg, Bing Xia, Marcelo A Carvalho, Alvaro N A Monteiro
{"title":"BRCA1 frameshift variants leading to extended incorrect protein C termini.","authors":"Thales C Nepomuceno, Tzeh Keong Foo, Marcy E Richardson, John Michael O Ranola, Jamie Weyandt, Matthew J Varga, Amaya Alarcon, Diana Gutierrez, Anna von Wachenfeldt, Daniel Eriksson, Raymond Kim, Susan Armel, Edwin Iversen, Fergus J Couch, Åke Borg, Bing Xia, Marcelo A Carvalho, Alvaro N A Monteiro","doi":"10.1016/j.xhgg.2023.100240","DOIUrl":null,"url":null,"abstract":"<p><p>Carriers of BRCA1 germline pathogenic variants are at substantially higher risk of developing breast and ovarian cancer than the general population. Accurate identification of at-risk individuals is crucial for risk stratification and the implementation of targeted preventive and therapeutic interventions. Despite significant progress in variant classification efforts, a sizable portion of reported BRCA1 variants remain as variants of uncertain clinical significance (VUSs). Variants leading to premature protein termination and loss of essential functional domains are typically classified as pathogenic. However, the impact of frameshift variants that result in an extended incorrect terminus is not clear. Using validated functional assays, we conducted a systematic functional assessment of 17 previously reported BRCA1 extended incorrect terminus variants (EITs) and concluded that 16 constitute loss-of-function variants. This suggests that most EITs are likely to be pathogenic. However, one variant, c.5578dup, displayed a protein expression level, affinity to known binding partners, and activity in transcription and homologous recombination assays comparable to the wild-type BRCA1 protein. Twenty-three additional carriers of c.5578dup were identified at a US clinical diagnostic lab and assessed using a family history likelihood model providing, in combination with the functional data, a likely benign interpretation. These results, consistent with family history data in the current study and available data from ClinVar, indicate that most, but not all, BRCA1 variants leading to an extended incorrect terminus constitute loss-of-function variants and underscore the need for comprehensive assessment of individual variants.</p>","PeriodicalId":34530,"journal":{"name":"HGG Advances","volume":" ","pages":"100240"},"PeriodicalIF":3.6000,"publicationDate":"2023-10-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e3/f9/main.PMC10558845.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HGG Advances","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.xhgg.2023.100240","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/9/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
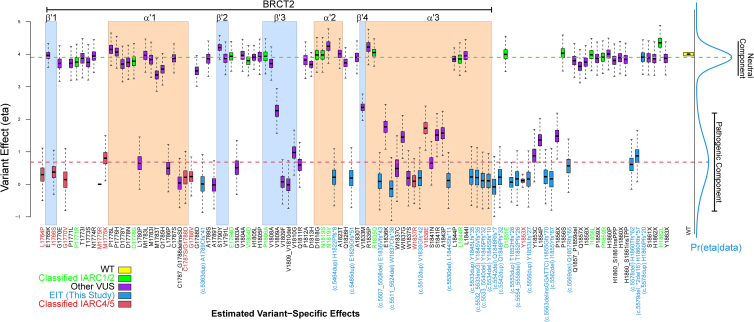
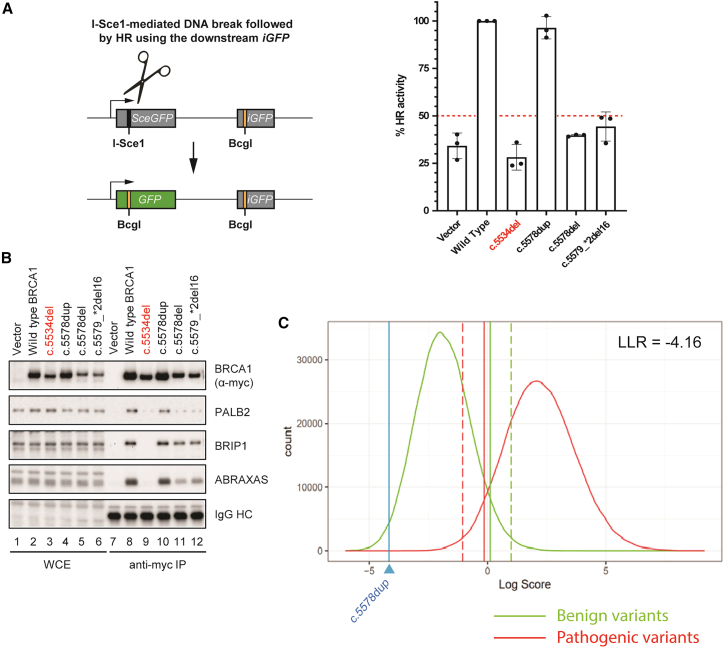
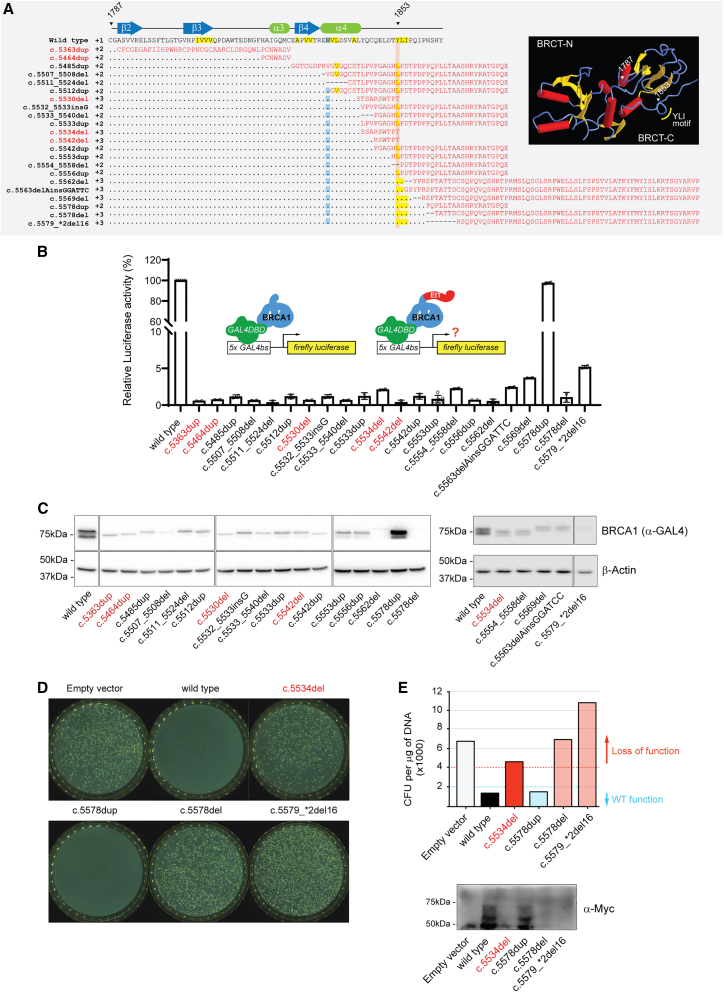
Carriers of BRCA1 germline pathogenic variants are at substantially higher risk of developing breast and ovarian cancer than the general population. Accurate identification of at-risk individuals is crucial for risk stratification and the implementation of targeted preventive and therapeutic interventions. Despite significant progress in variant classification efforts, a sizable portion of reported BRCA1 variants remain as variants of uncertain clinical significance (VUSs). Variants leading to premature protein termination and loss of essential functional domains are typically classified as pathogenic. However, the impact of frameshift variants that result in an extended incorrect terminus is not clear. Using validated functional assays, we conducted a systematic functional assessment of 17 previously reported BRCA1 extended incorrect terminus variants (EITs) and concluded that 16 constitute loss-of-function variants. This suggests that most EITs are likely to be pathogenic. However, one variant, c.5578dup, displayed a protein expression level, affinity to known binding partners, and activity in transcription and homologous recombination assays comparable to the wild-type BRCA1 protein. Twenty-three additional carriers of c.5578dup were identified at a US clinical diagnostic lab and assessed using a family history likelihood model providing, in combination with the functional data, a likely benign interpretation. These results, consistent with family history data in the current study and available data from ClinVar, indicate that most, but not all, BRCA1 variants leading to an extended incorrect terminus constitute loss-of-function variants and underscore the need for comprehensive assessment of individual variants.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
