{"title":"UMOD and you! Explaining a rare disease diagnosis.","authors":"Holly Mabillard, Eric Olinger, John A Sayer","doi":"10.1007/s44162-022-00005-4","DOIUrl":null,"url":null,"abstract":"<p><p>The precise molecular genetic diagnosis of a rare inherited disease is nearly always a prolonged odyssey. Fortunately, modern molecular testing strategies are allowing more diagnoses to be made. There are many different rare inherited kidney diseases and both the genetic heterogeneity of these conditions and the clinical diversity often leads to confusing nomenclature. Autosomal dominant tubulointerstitial kidney disease (ADTKD) is an example of this. ADTKD, an inherited kidney disease that leads to worsening of kidney function over time, often culminating in end stage kidney disease, accounting for around 2% of this cohort. <i>UMOD</i> is the most common gene implicated in this disorder but there are at least 6 subtypes. At present, there are no specific treatments for ADTKD. Here, we review the current understanding of this condition and provide patient-centred information to allow conceptual understanding of this disease to allow better recognition, diagnosis and management.</p>","PeriodicalId":73925,"journal":{"name":"Journal of rare diseases (Berlin, Germany)","volume":"1 1","pages":"4"},"PeriodicalIF":0.0000,"publicationDate":"2022-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9767401/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of rare diseases (Berlin, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s44162-022-00005-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2022/12/7 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
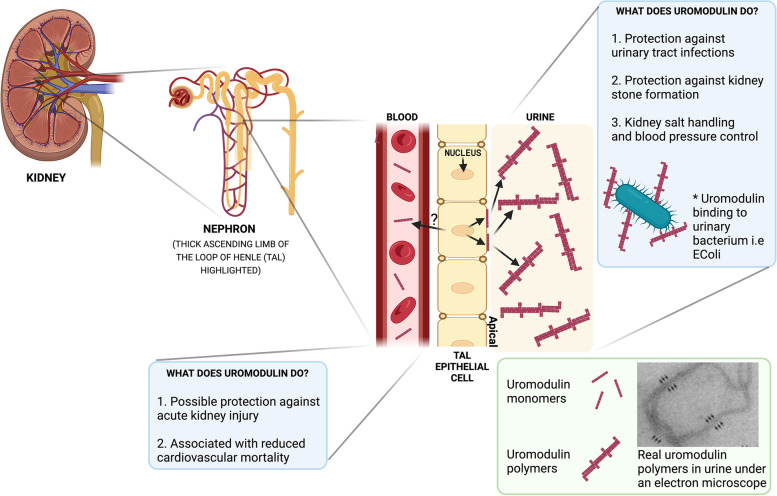
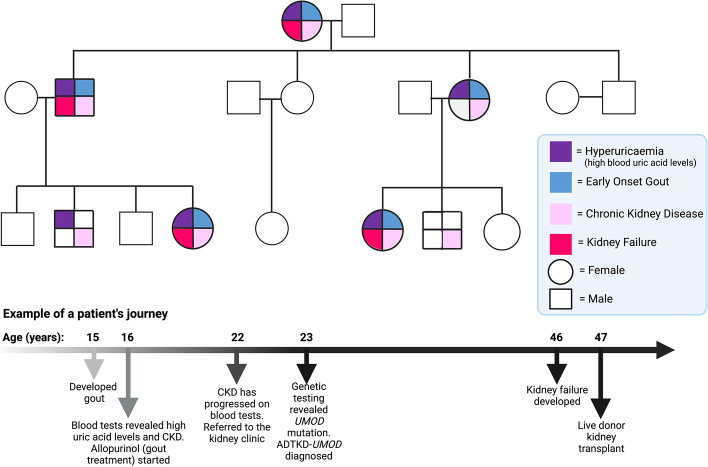
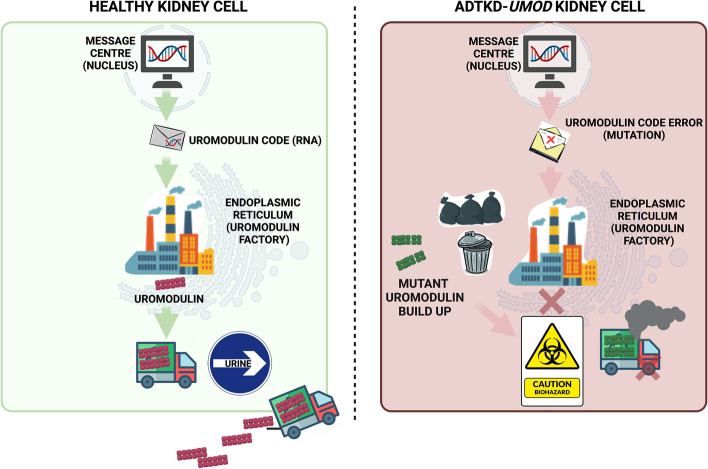
The precise molecular genetic diagnosis of a rare inherited disease is nearly always a prolonged odyssey. Fortunately, modern molecular testing strategies are allowing more diagnoses to be made. There are many different rare inherited kidney diseases and both the genetic heterogeneity of these conditions and the clinical diversity often leads to confusing nomenclature. Autosomal dominant tubulointerstitial kidney disease (ADTKD) is an example of this. ADTKD, an inherited kidney disease that leads to worsening of kidney function over time, often culminating in end stage kidney disease, accounting for around 2% of this cohort. UMOD is the most common gene implicated in this disorder but there are at least 6 subtypes. At present, there are no specific treatments for ADTKD. Here, we review the current understanding of this condition and provide patient-centred information to allow conceptual understanding of this disease to allow better recognition, diagnosis and management.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
