Profiling SARS-CoV-2 Main Protease (MPRO) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields
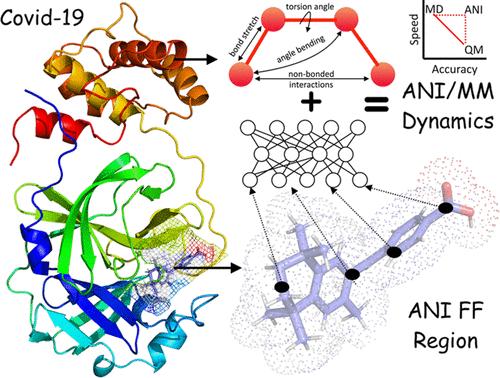
{"title":"Profiling SARS-CoV-2 Main Protease (MPRO) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields","authors":"Aayush Gupta, Huan-Xiang Zhou*","doi":"10.1021/acscombsci.0c00140","DOIUrl":null,"url":null,"abstract":"<p >The current COVID-19 pandemic caused by a novel coronavirus SARS-CoV-2 urgently calls for a working therapeutic. Here, we report a computation-based workflow for efficiently selecting a subset of FDA-approved drugs that can potentially bind to the SARS-CoV-2 main protease M<sup>PRO</sup>. The workflow started with docking (using Autodock Vina) each of 1615 FDA-approved drugs to the M<sup>PRO</sup> active site. This step selected 62 candidates with docking energies lower than ?8.5 kcal/mol. Then, the 62 docked protein–drug complexes were subjected to 100 ns of molecular dynamics (MD) simulations in a molecular mechanics (MM) force field (CHARMM36). This step reduced the candidate pool to 26, based on the root-mean-square-deviations (RMSDs) of the drug molecules in the trajectories. Finally, we modeled the 26 drug molecules by a pseudoquantum mechanical (ANI) force field and ran 5 ns hybrid ANI/MM MD simulations of the 26 protein–drug complexes. ANI was trained by neural network models on quantum mechanical density functional theory (wB97X/6-31G(d)) data points. An RMSD cutoff winnowed down the pool to 12, and free energy analysis (MM/PBSA) produced the final selection of 9 drugs: dihydroergotamine, midostaurin, ziprasidone, etoposide, apixaban, fluorescein, tadalafil, rolapitant, and palbociclib. Of these, three are found to be active in literature reports of experimental studies. To provide physical insight into their mechanism of action, the interactions of the drug molecules with the protein are presented as 2D-interaction maps. These findings and mappings of drug–protein interactions may be potentially used to guide rational drug discovery against COVID-19.</p>","PeriodicalId":14,"journal":{"name":"ACS Combinatorial Science","volume":"22 12","pages":"826–832"},"PeriodicalIF":3.7840,"publicationDate":"2020-10-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1021/acscombsci.0c00140","citationCount":"24","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Combinatorial Science","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscombsci.0c00140","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 24
Abstract
The current COVID-19 pandemic caused by a novel coronavirus SARS-CoV-2 urgently calls for a working therapeutic. Here, we report a computation-based workflow for efficiently selecting a subset of FDA-approved drugs that can potentially bind to the SARS-CoV-2 main protease MPRO. The workflow started with docking (using Autodock Vina) each of 1615 FDA-approved drugs to the MPRO active site. This step selected 62 candidates with docking energies lower than ?8.5 kcal/mol. Then, the 62 docked protein–drug complexes were subjected to 100 ns of molecular dynamics (MD) simulations in a molecular mechanics (MM) force field (CHARMM36). This step reduced the candidate pool to 26, based on the root-mean-square-deviations (RMSDs) of the drug molecules in the trajectories. Finally, we modeled the 26 drug molecules by a pseudoquantum mechanical (ANI) force field and ran 5 ns hybrid ANI/MM MD simulations of the 26 protein–drug complexes. ANI was trained by neural network models on quantum mechanical density functional theory (wB97X/6-31G(d)) data points. An RMSD cutoff winnowed down the pool to 12, and free energy analysis (MM/PBSA) produced the final selection of 9 drugs: dihydroergotamine, midostaurin, ziprasidone, etoposide, apixaban, fluorescein, tadalafil, rolapitant, and palbociclib. Of these, three are found to be active in literature reports of experimental studies. To provide physical insight into their mechanism of action, the interactions of the drug molecules with the protein are presented as 2D-interaction maps. These findings and mappings of drug–protein interactions may be potentially used to guide rational drug discovery against COVID-19.
期刊介绍:
The Journal of Combinatorial Chemistry has been relaunched as ACS Combinatorial Science under the leadership of new Editor-in-Chief M.G. Finn of The Scripps Research Institute. The journal features an expanded scope and will build upon the legacy of the Journal of Combinatorial Chemistry, a highly cited leader in the field.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
