Narendra Babu Kondapalli , Venkatesh Katari , Kesha Dalal, Sailaja Paruchuri, Charles K. Thodeti
{"title":"Angiotensin II induces endothelial dysfunction and vascular remodeling by downregulating TRPV4 channels","authors":"Narendra Babu Kondapalli , Venkatesh Katari , Kesha Dalal, Sailaja Paruchuri, Charles K. Thodeti","doi":"10.1016/j.jmccpl.2023.100055","DOIUrl":null,"url":null,"abstract":"<div><p>Angiotensin II (Ang II) is a potent vasoconstrictor of vascular smooth muscle cells (VSMC) and is implicated in hypertension, but it's role in the regulation of endothelial function is not well known. We and others have previously shown that mechanically activated ion channel, Transient Receptor Potential Vanilloid 4 (TRPV4) mediates flow- and/or receptor-dependent vasodilation via nitric oxide (NO) production in endothelial cells. Ang II was demonstrated to crosstalk with TRPV4 via angiotensin 1 receptor (AT1R) and β-arrestin signaling in epithelial and immortalized cells, however, the role of this crosstalk in endothelial cell function is not fully explored. Ang II treatment significantly downregulated TRPV4 protein expression and TRPV4-mediated Ca<sup>2+</sup> influx in human EC without altering TRPV4 mRNA levels. Further, TRPV4-induced eNOS phosphorylation and NO production were significantly reduced in Ang II-treated human EC. Importantly, Ang II infusion in mice revealed that, TRPV4/p-eNOS expression and colocalization was reduced in endothelium in vivo. Finally, Ang II infusion induced vascular remodeling as evidenced by decreased lumen to wall ratio in resistant mesenteric arteries. These findings suggest that Ang II induces endothelial dysfunction and vascular remodeling via downregulation of TRPV4/eNOS pathway and may contribute to hypertension, independent of or in addition to its effect on vascular smooth muscle contraction.</p></div>","PeriodicalId":73835,"journal":{"name":"Journal of molecular and cellular cardiology plus","volume":"6 ","pages":"Article 100055"},"PeriodicalIF":2.2000,"publicationDate":"2023-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2772976123000259/pdfft?md5=328dd4ebe56f8de016e69018e4ecf08e&pid=1-s2.0-S2772976123000259-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology plus","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772976123000259","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
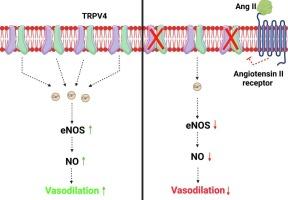
Angiotensin II (Ang II) is a potent vasoconstrictor of vascular smooth muscle cells (VSMC) and is implicated in hypertension, but it's role in the regulation of endothelial function is not well known. We and others have previously shown that mechanically activated ion channel, Transient Receptor Potential Vanilloid 4 (TRPV4) mediates flow- and/or receptor-dependent vasodilation via nitric oxide (NO) production in endothelial cells. Ang II was demonstrated to crosstalk with TRPV4 via angiotensin 1 receptor (AT1R) and β-arrestin signaling in epithelial and immortalized cells, however, the role of this crosstalk in endothelial cell function is not fully explored. Ang II treatment significantly downregulated TRPV4 protein expression and TRPV4-mediated Ca2+ influx in human EC without altering TRPV4 mRNA levels. Further, TRPV4-induced eNOS phosphorylation and NO production were significantly reduced in Ang II-treated human EC. Importantly, Ang II infusion in mice revealed that, TRPV4/p-eNOS expression and colocalization was reduced in endothelium in vivo. Finally, Ang II infusion induced vascular remodeling as evidenced by decreased lumen to wall ratio in resistant mesenteric arteries. These findings suggest that Ang II induces endothelial dysfunction and vascular remodeling via downregulation of TRPV4/eNOS pathway and may contribute to hypertension, independent of or in addition to its effect on vascular smooth muscle contraction.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
