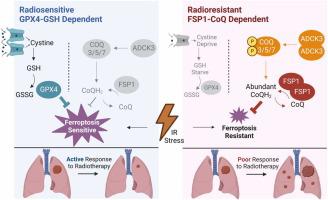
Acquired radioresistance is the primary contributor to treatment failure of radiotherapy, with ferroptosis is identified as a significant mechanism underlying cell death during radiotherapy. Although resistance to ferroptosis has been observed in both clinical samples of radioresistant cells and cell models, its mechanism remains unidentified. Herein, our investigation revealed that radioresistant cells exhibited greater tolerance to Glutathione Peroxidase 4 (GPX4) inhibitors and, conversely, increased sensitivity to ferroptosis suppressor protein 1 (FSP1) inhibitors compared to their sensitive counterparts. This observation suggested that FSP1 might play a dominant role in the development of radioresistance. Notably, the knockout of FSP1 demonstrated considerably superior efficacy in resensitizing cells to radiotherapy compared to the knockout of GPX4. To elucidate the driving force behind this functional shift, we conducted a metabolomic assay, which revealed an upregulation of Coenzyme Q (CoQ) synthesis and a downregulation of glutathione synthesis in the acquired radioresistance cells. Mechanistically, CoQ synthesis was found to be supported by aarF domain containing kinase 3-mediated phosphorylation of CoQ synthases, while the downregulation of Solute carrier family 7 member 11 led to decreased glutathione synthesis. Remarkably, our retrospective analysis of clinical response data further validated that the additional administration of statin during radiotherapy, which could impede CoQ production, effectively resensitized radioresistant cells to radiation. In summary, our findings demonstrate a dependency shift from GPX4 to FSP1 driven by altered metabolite synthesis during the acquisition of radioresistance. Moreover, we provide a promising therapeutic strategy for reversing radioresistance by inhibiting the FSP1-CoQ pathway.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
