Molecular docking studies of some benzoxazole and benzothiazole derivatives as VEGFR-2 target inhibitors: In silico design, MD simulation, pharmacokinetics and DFT studies
Sagiru Hamza Abdullahi , Abu Tayab Moin , Adamu Uzairu , Abdullahi Bello Umar , Muhammad Tukur Ibrahim , Mustapha Tijjani Usman , Nafisa Nawal , Imren Bayil , Talha Zubair
{"title":"Molecular docking studies of some benzoxazole and benzothiazole derivatives as VEGFR-2 target inhibitors: In silico design, MD simulation, pharmacokinetics and DFT studies","authors":"Sagiru Hamza Abdullahi , Abu Tayab Moin , Adamu Uzairu , Abdullahi Bello Umar , Muhammad Tukur Ibrahim , Mustapha Tijjani Usman , Nafisa Nawal , Imren Bayil , Talha Zubair","doi":"10.1016/j.ipha.2023.11.010","DOIUrl":null,"url":null,"abstract":"<div><p>Breast cancer, a deadly disease among women, demands effective interventions due to its global impact, with over one million annual cases. Current anti-breast cancer drugs displays several side effects, also most patients resists to these drugs during early treatment stage. Hence, global search for better drugs with less side effects became necessary. The objective of this study is to identify potential inhibitors of vascular endothelial growth factor receptor-2 (VEGFR-2), a critical target in breast cancer treatment. Molecular docking-based virtual screening of 45 benzoxazole/thiazole derivatives was conducted, followed by molecular dynamics simulations to explore ligand-protein interactions. Seven ligands (compounds 7, 10, 12, 13, 14, 20, and 26) demonstrated superior binding affinities ranging from −157.85 to −173.88 kcal/mol (MolDock scores) and −109.96 to −129.23 kcal/mol (Re-rank scores) compared to Sorafenib (−156.35 kcal/mol MolDock score and −102.63 kcal/mol Re-rank score). Compound 7, identified as a potential hit, exhibited stability in a 100-ns dynamic simulation. It was chosen as a template for designing novel inhibitors, resulting in five compounds with improved binding affinities ranging from −177.84 to −184.69 kcal/mol (MolDock scores) and −137.34 to −143.44 kcal/mol (Re-rank scores). Pharmacological profiling confirmed the drug-like properties of both the potential hit molecules and the designed compounds, with 0–1 violations against Lipinski's rule of five and favorable pharmacokinetics status. Density functional theory (DFT) studies illustrated the reactive nature of the designed compounds. These findings suggest the potential of these molecules as novel VEGFR-2 inhibitors for breast cancer treatment, providing promising prospects for future drug development.</p></div>","PeriodicalId":100682,"journal":{"name":"Intelligent Pharmacy","volume":"2 2","pages":"Pages 232-250"},"PeriodicalIF":0.0000,"publicationDate":"2023-12-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2949866X23001247/pdfft?md5=deba183193dcd5333b7237f57dbc3641&pid=1-s2.0-S2949866X23001247-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Intelligent Pharmacy","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2949866X23001247","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
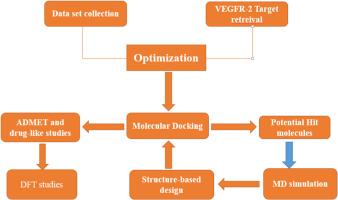
Breast cancer, a deadly disease among women, demands effective interventions due to its global impact, with over one million annual cases. Current anti-breast cancer drugs displays several side effects, also most patients resists to these drugs during early treatment stage. Hence, global search for better drugs with less side effects became necessary. The objective of this study is to identify potential inhibitors of vascular endothelial growth factor receptor-2 (VEGFR-2), a critical target in breast cancer treatment. Molecular docking-based virtual screening of 45 benzoxazole/thiazole derivatives was conducted, followed by molecular dynamics simulations to explore ligand-protein interactions. Seven ligands (compounds 7, 10, 12, 13, 14, 20, and 26) demonstrated superior binding affinities ranging from −157.85 to −173.88 kcal/mol (MolDock scores) and −109.96 to −129.23 kcal/mol (Re-rank scores) compared to Sorafenib (−156.35 kcal/mol MolDock score and −102.63 kcal/mol Re-rank score). Compound 7, identified as a potential hit, exhibited stability in a 100-ns dynamic simulation. It was chosen as a template for designing novel inhibitors, resulting in five compounds with improved binding affinities ranging from −177.84 to −184.69 kcal/mol (MolDock scores) and −137.34 to −143.44 kcal/mol (Re-rank scores). Pharmacological profiling confirmed the drug-like properties of both the potential hit molecules and the designed compounds, with 0–1 violations against Lipinski's rule of five and favorable pharmacokinetics status. Density functional theory (DFT) studies illustrated the reactive nature of the designed compounds. These findings suggest the potential of these molecules as novel VEGFR-2 inhibitors for breast cancer treatment, providing promising prospects for future drug development.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
