Emrah Gumusgoz, Sahba Kasiri, Mayank Verma, Jun Wu, Daniel Villarreal Acha, Ummay Marriam, Sharyl Fyffe-Maricich, Amy Lin, Xin Chen, Steven J. Gray, Berge A. Minassian
{"title":"CSTB gene replacement improves neuroinflammation, neurodegeneration and ataxia in murine type 1 progressive myoclonus epilepsy","authors":"Emrah Gumusgoz, Sahba Kasiri, Mayank Verma, Jun Wu, Daniel Villarreal Acha, Ummay Marriam, Sharyl Fyffe-Maricich, Amy Lin, Xin Chen, Steven J. Gray, Berge A. Minassian","doi":"10.1038/s41434-023-00433-x","DOIUrl":null,"url":null,"abstract":"EPM1 is the most common form of Progressive Myoclonus Epilepsy characterized by late-childhood onset, ever-worsening and disabling myoclonus, seizures, ataxia, psychiatric disease, and shortened lifespan. EPM1 is caused by expansions of a dodecamer repeat sequence in the promoter of CSTB (cystatin B), which dramatically reduces, but does not eliminate, gene expression. The relatively late onset and consistent presence of a minimal amount of protein product makes EPM1 a favorable target for gene replacement therapy. If treated early, these children’s normally developed brains could be rescued from the neurodegeneration that otherwise follows, and their cross-reactive immunological material (CRIM) positive status greatly reduces transgene related toxicity. We performed a proof-of-concept CSTB gene replacement study in Cstb knockout mice by introducing full-length human CSTB driven by the CBh promoter packaged in AAV9 and administered at postnatal days 21 and 60. Mice were sacrificed at 2 or 9 months of age, respectively. We observed significant improvements in expression levels of neuroinflammatory pathway genes and cerebellar granule cell layer apoptosis, as well as amelioration of motor impairment. The data suggest that gene replacement is a promising therapeutic modality for EPM1 and could spare affected children and families the ravages of this otherwise severe neurodegenerative disease.","PeriodicalId":12699,"journal":{"name":"Gene Therapy","volume":"31 5-6","pages":"234-241"},"PeriodicalIF":4.5000,"publicationDate":"2023-12-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Gene Therapy","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s41434-023-00433-x","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
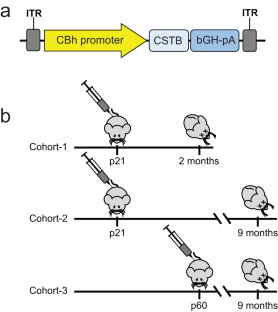
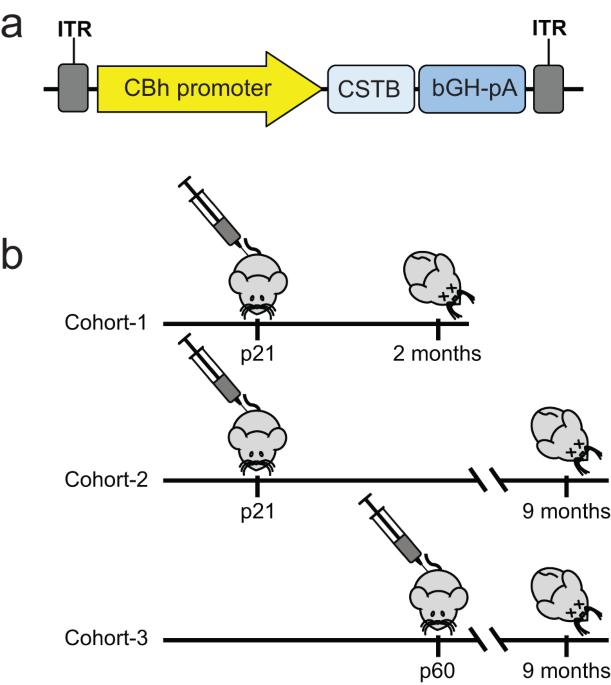
EPM1 is the most common form of Progressive Myoclonus Epilepsy characterized by late-childhood onset, ever-worsening and disabling myoclonus, seizures, ataxia, psychiatric disease, and shortened lifespan. EPM1 is caused by expansions of a dodecamer repeat sequence in the promoter of CSTB (cystatin B), which dramatically reduces, but does not eliminate, gene expression. The relatively late onset and consistent presence of a minimal amount of protein product makes EPM1 a favorable target for gene replacement therapy. If treated early, these children’s normally developed brains could be rescued from the neurodegeneration that otherwise follows, and their cross-reactive immunological material (CRIM) positive status greatly reduces transgene related toxicity. We performed a proof-of-concept CSTB gene replacement study in Cstb knockout mice by introducing full-length human CSTB driven by the CBh promoter packaged in AAV9 and administered at postnatal days 21 and 60. Mice were sacrificed at 2 or 9 months of age, respectively. We observed significant improvements in expression levels of neuroinflammatory pathway genes and cerebellar granule cell layer apoptosis, as well as amelioration of motor impairment. The data suggest that gene replacement is a promising therapeutic modality for EPM1 and could spare affected children and families the ravages of this otherwise severe neurodegenerative disease.
期刊介绍:
Gene Therapy covers both the research and clinical applications of novel therapeutic techniques based on a genetic component. Over the last few decades, significant advances in technologies ranging from identifying novel genetic targets that cause disease through to clinical studies, which show therapeutic benefit, have elevated this multidisciplinary field to the forefront of modern medicine.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
