{"title":"Cryo-forum: A framework for orientation recovery with uncertainty measure with the application in cryo-EM image analysis","authors":"Szu-Chi Chung","doi":"10.1016/j.jsb.2023.108058","DOIUrl":null,"url":null,"abstract":"<div><p>In single-particle cryo-electron microscopy (cryo-EM), efficient determination of orientation parameters for particle images poses a significant challenge yet is crucial for reconstructing 3D structures. This task is complicated by the high noise levels in the datasets, which often include outliers, necessitating several time-consuming 2D clean-up processes. Recently, solutions based on deep learning have emerged, offering a more streamlined approach to the traditionally laborious task of orientation estimation. These solutions employ amortized inference, eliminating the need to estimate parameters individually for each image. However, these methods frequently overlook the presence of outliers and may not adequately concentrate on the components used within the network. This paper introduces a novel method using a 10-dimensional feature vector for orientation representation, extracting orientations as unit quaternions with an accompanying uncertainty metric. Furthermore, we propose a unique loss function that considers the pairwise distances between orientations, thereby enhancing the accuracy of our method. Finally, we also comprehensively evaluate the design choices in constructing the encoder network, a topic that has not received sufficient attention in the literature. Our numerical analysis demonstrates that our methodology effectively recovers orientations from 2D cryo-EM images in an end-to-end manner. Notably, the inclusion of uncertainty quantification allows for direct clean-up of the dataset at the 3D level. Lastly, we package our proposed methods into a user-friendly software suite named <em>cryo-forum</em>, designed for easy access by developers.</p></div>","PeriodicalId":17074,"journal":{"name":"Journal of structural biology","volume":"216 1","pages":"Article 108058"},"PeriodicalIF":2.7000,"publicationDate":"2024-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of structural biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1047847723001211","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/12/30 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
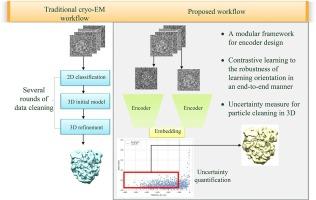
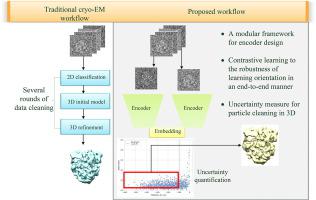
In single-particle cryo-electron microscopy (cryo-EM), efficient determination of orientation parameters for particle images poses a significant challenge yet is crucial for reconstructing 3D structures. This task is complicated by the high noise levels in the datasets, which often include outliers, necessitating several time-consuming 2D clean-up processes. Recently, solutions based on deep learning have emerged, offering a more streamlined approach to the traditionally laborious task of orientation estimation. These solutions employ amortized inference, eliminating the need to estimate parameters individually for each image. However, these methods frequently overlook the presence of outliers and may not adequately concentrate on the components used within the network. This paper introduces a novel method using a 10-dimensional feature vector for orientation representation, extracting orientations as unit quaternions with an accompanying uncertainty metric. Furthermore, we propose a unique loss function that considers the pairwise distances between orientations, thereby enhancing the accuracy of our method. Finally, we also comprehensively evaluate the design choices in constructing the encoder network, a topic that has not received sufficient attention in the literature. Our numerical analysis demonstrates that our methodology effectively recovers orientations from 2D cryo-EM images in an end-to-end manner. Notably, the inclusion of uncertainty quantification allows for direct clean-up of the dataset at the 3D level. Lastly, we package our proposed methods into a user-friendly software suite named cryo-forum, designed for easy access by developers.
期刊介绍:
Journal of Structural Biology (JSB) has an open access mirror journal, the Journal of Structural Biology: X (JSBX), sharing the same aims and scope, editorial team, submission system and rigorous peer review. Since both journals share the same editorial system, you may submit your manuscript via either journal homepage. You will be prompted during submission (and revision) to choose in which to publish your article. The editors and reviewers are not aware of the choice you made until the article has been published online. JSB and JSBX publish papers dealing with the structural analysis of living material at every level of organization by all methods that lead to an understanding of biological function in terms of molecular and supermolecular structure.
Techniques covered include:
• Light microscopy including confocal microscopy
• All types of electron microscopy
• X-ray diffraction
• Nuclear magnetic resonance
• Scanning force microscopy, scanning probe microscopy, and tunneling microscopy
• Digital image processing
• Computational insights into structure




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
