Keighlynn A. Veilleux, Georg Schreckenbach and David E. Herbert
{"title":"Designing biphenanthridine-based singlet fission materials using computational chemistry†","authors":"Keighlynn A. Veilleux, Georg Schreckenbach and David E. Herbert","doi":"10.1039/D3ME00181D","DOIUrl":null,"url":null,"abstract":"<p >Singlet fission has the potential to significantly improve the efficiency of photovoltaic devices by harnessing high-energy sunlight to double the photocurrent that can be generated in standard semiconductors. The challenge is identifying materials capable of undergoing this process efficiently. Herein, we present the results of a systematic search for novel intermolecular singlet fission materials based on the recently synthesized 6,6′-biphenanthridine (biphe) framework utilizing a straightforward computational approach. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were employed to study the photophysics of various structural analogues of biphe. These analogues were generated <em>in silico</em> by utilizing an extensive range of transformations, including planarization, protonation, symmetric and asymmetric alkylation, electron-donating and electron-withdrawing group substitution, <em>N</em>-oxide substitution, and symmetric and asymmetric π-extension and contraction. Analysis of the effects of these structural modifications on the energies of the lowest singlet and triplet excited states revealed that (2,2′,10,10′-tetra-<em>N</em>-oxide) planar biphe has an <em>E</em>(S<small><sub>1</sub></small>)/<em>E</em>(T<small><sub>1</sub></small>) ratio of 2.12 and an <em>E</em>(T<small><sub>2</sub></small>)/<em>E</em>(T<small><sub>1</sub></small>) of 2.05, suggesting its potential for intermolecular singlet fission. Additionally, <em>N</em>-methylated biphe emerged as a promising contender for thermally activated delayed fluorescence. The effects of solvation are also discussed.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 4","pages":" 423-435"},"PeriodicalIF":3.2000,"publicationDate":"2024-02-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d3me00181d","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
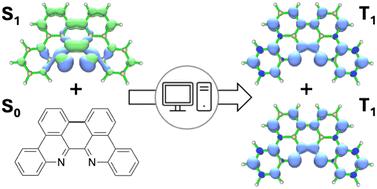
Singlet fission has the potential to significantly improve the efficiency of photovoltaic devices by harnessing high-energy sunlight to double the photocurrent that can be generated in standard semiconductors. The challenge is identifying materials capable of undergoing this process efficiently. Herein, we present the results of a systematic search for novel intermolecular singlet fission materials based on the recently synthesized 6,6′-biphenanthridine (biphe) framework utilizing a straightforward computational approach. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) were employed to study the photophysics of various structural analogues of biphe. These analogues were generated in silico by utilizing an extensive range of transformations, including planarization, protonation, symmetric and asymmetric alkylation, electron-donating and electron-withdrawing group substitution, N-oxide substitution, and symmetric and asymmetric π-extension and contraction. Analysis of the effects of these structural modifications on the energies of the lowest singlet and triplet excited states revealed that (2,2′,10,10′-tetra-N-oxide) planar biphe has an E(S1)/E(T1) ratio of 2.12 and an E(T2)/E(T1) of 2.05, suggesting its potential for intermolecular singlet fission. Additionally, N-methylated biphe emerged as a promising contender for thermally activated delayed fluorescence. The effects of solvation are also discussed.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
