{"title":"From single Cu atoms to sub-nanometric copper clusters deposited on TiO2: a DFT study","authors":"Dorota Rutkowska-Zbik, Agnieszka Drzewiecka-Matuszek, Renata Tokarz-Sobieraj","doi":"10.1007/s11224-024-02288-3","DOIUrl":null,"url":null,"abstract":"<div><p>The growing interest in a material composed of Cu single atoms and/or their sub-nanometric clusters deposited on titania prompted us to perform systematic theoretical studies on the system comprising the anatase phase of titania, modelled by a (TiO<sub>2</sub>)<sub>34</sub> cluster with copper particles of 1–7 atoms on top of it. The ground-state geometric structures were proposed and compared with the available literature data derived from EXAFS experiments done for Cu-TiO<sub>2</sub> materials. Copper atoms prefer to aggregate and form larger clusters on TiO<sub>2</sub>, as seen from the computed nucleation energies. The models were characterised by the following electronic properties: electronic band structure, natural population charges, and frontier orbitals (HOMO, LUMO, and SOMO). The copper phase becomes oxidised once it is deposited on titania. The charge distribution in the resulting structures indicates that the atoms that are the closest to the Cu-TiO<sub>2</sub> interface would become the active sites for catalytic processes; copper atoms would act as electrophilic, while oxygen atoms would act as nucleophilic. The calculated binding energies between the two phases show that the formation of the composite system is favourable from the thermodynamic point of view, and the interaction between the small copper clusters and the titania surface is mostly of electrostatic nature.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"35 5","pages":"1449 - 1459"},"PeriodicalIF":2.2000,"publicationDate":"2024-02-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://link.springer.com/content/pdf/10.1007/s11224-024-02288-3.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02288-3","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
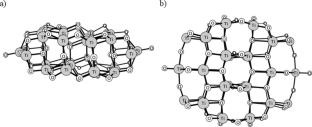
The growing interest in a material composed of Cu single atoms and/or their sub-nanometric clusters deposited on titania prompted us to perform systematic theoretical studies on the system comprising the anatase phase of titania, modelled by a (TiO2)34 cluster with copper particles of 1–7 atoms on top of it. The ground-state geometric structures were proposed and compared with the available literature data derived from EXAFS experiments done for Cu-TiO2 materials. Copper atoms prefer to aggregate and form larger clusters on TiO2, as seen from the computed nucleation energies. The models were characterised by the following electronic properties: electronic band structure, natural population charges, and frontier orbitals (HOMO, LUMO, and SOMO). The copper phase becomes oxidised once it is deposited on titania. The charge distribution in the resulting structures indicates that the atoms that are the closest to the Cu-TiO2 interface would become the active sites for catalytic processes; copper atoms would act as electrophilic, while oxygen atoms would act as nucleophilic. The calculated binding energies between the two phases show that the formation of the composite system is favourable from the thermodynamic point of view, and the interaction between the small copper clusters and the titania surface is mostly of electrostatic nature.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
