{"title":"Cipaglucosidase alfa-atga: Unveiling new horizons in Pompe disease therapy","authors":"Arshdeep Singh , Rabin Debnath , Aniket Saini , Kushal Seni , Anjali Sharma , Deepak Singh Bisht , Viney Chawla , Pooja A Chawla","doi":"10.1016/j.hsr.2024.100160","DOIUrl":null,"url":null,"abstract":"<div><p>Pompe disease is a lysosomal storage disease characterized by impaired glycogen breakdown due to an acid α-glucosidase (GAA) enzyme deficiency. Without therapy, children with the severe infantile form do not survive past their first year of life. POMBILITI which is intended to treat late-onset Pompe disease. The enzyme cipaglucosidase alfa-atga, which is produced from Chinese Hamster Ovary (CHO) cells, is a component of this novel drug. This enzyme is produced using a highly developed process known as perfusion methodology. Recombinant human α-glucosidase (rhGAA) is expressed and produced in CHO cells using the perfusion process. This drug helps to treat Pompe disease by the breakdown of glycogen within lysosomes. Late-onset Pompe disease is characterized by a deficiency in Alpha glucosidase, leading to the accumulation of glycogen within lysosomes and subsequent cellular dysfunction. POMBILITI's targeted approach involves the administration of the rhGAA enzyme, providing a therapeutic replacement for the deficient natural enzyme. This drug aims to restore the normal physiological function of lysosomes, thereby mitigating the impact of Pompe disease on affected individuals. The current study is focused on the drug cipaglucosidase alfa-atga which the FDA has approved for the treatment of Pompe disease on 28 September 2023.</p></div>","PeriodicalId":73214,"journal":{"name":"Health sciences review (Oxford, England)","volume":"11 ","pages":"Article 100160"},"PeriodicalIF":0.0000,"publicationDate":"2024-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2772632024000138/pdfft?md5=3bdff161a233a6477caa05ad78ea5831&pid=1-s2.0-S2772632024000138-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Health sciences review (Oxford, England)","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772632024000138","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
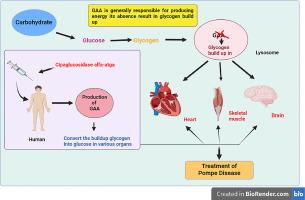
Pompe disease is a lysosomal storage disease characterized by impaired glycogen breakdown due to an acid α-glucosidase (GAA) enzyme deficiency. Without therapy, children with the severe infantile form do not survive past their first year of life. POMBILITI which is intended to treat late-onset Pompe disease. The enzyme cipaglucosidase alfa-atga, which is produced from Chinese Hamster Ovary (CHO) cells, is a component of this novel drug. This enzyme is produced using a highly developed process known as perfusion methodology. Recombinant human α-glucosidase (rhGAA) is expressed and produced in CHO cells using the perfusion process. This drug helps to treat Pompe disease by the breakdown of glycogen within lysosomes. Late-onset Pompe disease is characterized by a deficiency in Alpha glucosidase, leading to the accumulation of glycogen within lysosomes and subsequent cellular dysfunction. POMBILITI's targeted approach involves the administration of the rhGAA enzyme, providing a therapeutic replacement for the deficient natural enzyme. This drug aims to restore the normal physiological function of lysosomes, thereby mitigating the impact of Pompe disease on affected individuals. The current study is focused on the drug cipaglucosidase alfa-atga which the FDA has approved for the treatment of Pompe disease on 28 September 2023.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
