S Zucca, G Nicora, F De Paoli, M G Carta, R Bellazzi, P Magni, E Rizzo, I Limongelli
{"title":"An AI-based approach driven by genotypes and phenotypes to uplift the diagnostic yield of genetic diseases.","authors":"S Zucca, G Nicora, F De Paoli, M G Carta, R Bellazzi, P Magni, E Rizzo, I Limongelli","doi":"10.1007/s00439-023-02638-x","DOIUrl":null,"url":null,"abstract":"<p><p>Identifying disease-causing variants in Rare Disease patients' genome is a challenging problem. To accomplish this task, we describe a machine learning framework, that we called \"Suggested Diagnosis\", whose aim is to prioritize genetic variants in an exome/genome based on the probability of being disease-causing. To do so, our method leverages standard guidelines for germline variant interpretation as defined by the American College of Human Genomics (ACMG) and the Association for Molecular Pathology (AMP), inheritance information, phenotypic similarity, and variant quality. Starting from (1) the VCF file containing proband's variants, (2) the list of proband's phenotypes encoded in Human Phenotype Ontology terms, and optionally (3) the information about family members (if available), the \"Suggested Diagnosis\" ranks all the variants according to their machine learning prediction. This method significantly reduces the number of variants that need to be evaluated by geneticists by pinpointing causative variants in the very first positions of the prioritized list. Most importantly, our approach proved to be among the top performers within the CAGI6 Rare Genome Project Challenge, where it was able to rank the true causative variant among the first positions and, uniquely among all the challenge participants, increased the diagnostic yield of 12.5% by solving 2 undiagnosed cases.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"159-171"},"PeriodicalIF":3.4000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11976766/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-023-02638-x","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/3/23 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
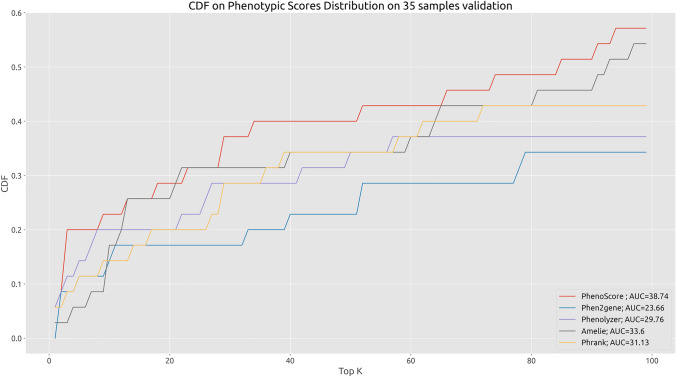
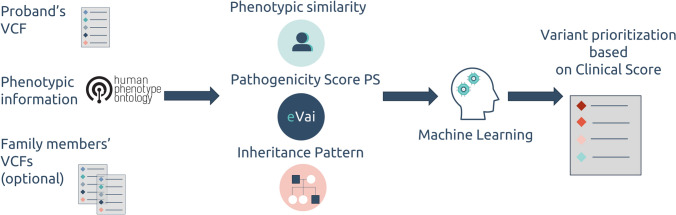
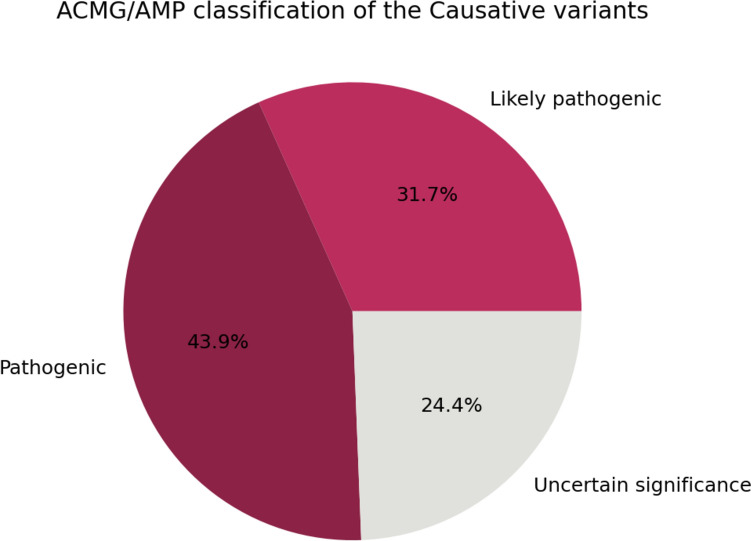
Identifying disease-causing variants in Rare Disease patients' genome is a challenging problem. To accomplish this task, we describe a machine learning framework, that we called "Suggested Diagnosis", whose aim is to prioritize genetic variants in an exome/genome based on the probability of being disease-causing. To do so, our method leverages standard guidelines for germline variant interpretation as defined by the American College of Human Genomics (ACMG) and the Association for Molecular Pathology (AMP), inheritance information, phenotypic similarity, and variant quality. Starting from (1) the VCF file containing proband's variants, (2) the list of proband's phenotypes encoded in Human Phenotype Ontology terms, and optionally (3) the information about family members (if available), the "Suggested Diagnosis" ranks all the variants according to their machine learning prediction. This method significantly reduces the number of variants that need to be evaluated by geneticists by pinpointing causative variants in the very first positions of the prioritized list. Most importantly, our approach proved to be among the top performers within the CAGI6 Rare Genome Project Challenge, where it was able to rank the true causative variant among the first positions and, uniquely among all the challenge participants, increased the diagnostic yield of 12.5% by solving 2 undiagnosed cases.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
