{"title":"Identification of hub genes and establishment of a diagnostic model in tuberculosis infection","authors":"Chunli Liu, Xing Li","doi":"10.1186/s13568-024-01691-7","DOIUrl":null,"url":null,"abstract":"<p>Tuberculosis (TB) poses significant challenges due to its high transmissibility within populations and intrinsic resistance to treatment, rendering it a formidable respiratory disease with a substantial susceptibility burden. This study was designed to identify new potential therapeutic targets for TB and establish a diagnostic model. mRNA expression data for TB were from GEO database, followed by conducting differential expression analysis. The top 50 genes with differential expression were subjected to GO and KEGG enrichment analyses. To establish a PPI network, the STRING database was utilized, and hub genes were identified utilizing five algorithms (EPC, MCC, MNC, Radiality, and Stress) within the cytoHubba plugin of Cytoscape software. Furthermore, a hub gene co-expression network was constructed using the GeneMANIA database. Consistency clustering was performed on hub genes, and ssGSEA was utilized to analyze the extent of immune infiltration in different subgroups. LASSO analysis was employed to construct a diagnostic model, and ROC curves were used for validation. Through the analysis of GEO data, a total of 159 genes were identified as differentially expressed. Further, GO and KEGG enrichment analyses revealed that these genes were mainly enriched in viral defense, symbiotic defense, and innate immune response-related pathways. Hub genes, including DDX58, IFIT2, IFIH1, RSAD2, IFI44L, OAS2, OAS1, OASL, IFIT1, IFIT3, MX1, STAT1, and ISG15, were identified using cytoHubba analysis of the PPI network. The GeneMANIA analysis unmasked that the co-expression rate of hub genes was 81.55%, and the physical interaction rate was 12.27%. Consistency clustering divided TB patients into two subgroups, and ssGSEA revealed different degrees of immune infiltration in different subgroups. LASSO analysis identified IFIT1, IFIT2, IFIT3, IFIH1, RSAD2, OAS1, OAS2, and STAT1 as eight immune-related key genes, and a diagnostic model was constructed. The ROC curve demonstrated that the model exhibited excellent diagnostic performance. DDX58, IFIT2, IFIH1, RSAD2, IFI44L, OAS2, OAS1, OASL, IFIT1, IFIT3, MX1, STAT1, and ISG15 were hub genes in TB, and the diagnostic model based on eight immune-related key genes exhibited good diagnostic performance.</p>","PeriodicalId":7537,"journal":{"name":"AMB Express","volume":"90 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2024-04-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"AMB Express","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.1186/s13568-024-01691-7","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
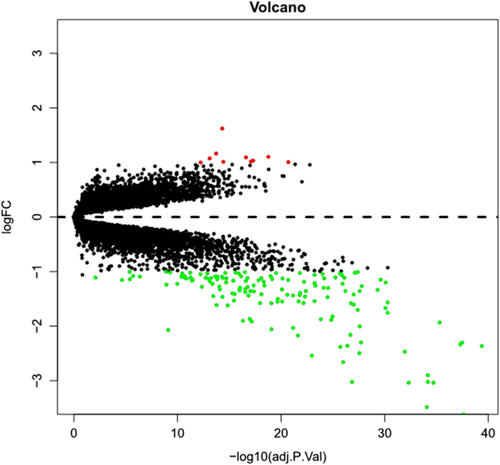
Tuberculosis (TB) poses significant challenges due to its high transmissibility within populations and intrinsic resistance to treatment, rendering it a formidable respiratory disease with a substantial susceptibility burden. This study was designed to identify new potential therapeutic targets for TB and establish a diagnostic model. mRNA expression data for TB were from GEO database, followed by conducting differential expression analysis. The top 50 genes with differential expression were subjected to GO and KEGG enrichment analyses. To establish a PPI network, the STRING database was utilized, and hub genes were identified utilizing five algorithms (EPC, MCC, MNC, Radiality, and Stress) within the cytoHubba plugin of Cytoscape software. Furthermore, a hub gene co-expression network was constructed using the GeneMANIA database. Consistency clustering was performed on hub genes, and ssGSEA was utilized to analyze the extent of immune infiltration in different subgroups. LASSO analysis was employed to construct a diagnostic model, and ROC curves were used for validation. Through the analysis of GEO data, a total of 159 genes were identified as differentially expressed. Further, GO and KEGG enrichment analyses revealed that these genes were mainly enriched in viral defense, symbiotic defense, and innate immune response-related pathways. Hub genes, including DDX58, IFIT2, IFIH1, RSAD2, IFI44L, OAS2, OAS1, OASL, IFIT1, IFIT3, MX1, STAT1, and ISG15, were identified using cytoHubba analysis of the PPI network. The GeneMANIA analysis unmasked that the co-expression rate of hub genes was 81.55%, and the physical interaction rate was 12.27%. Consistency clustering divided TB patients into two subgroups, and ssGSEA revealed different degrees of immune infiltration in different subgroups. LASSO analysis identified IFIT1, IFIT2, IFIT3, IFIH1, RSAD2, OAS1, OAS2, and STAT1 as eight immune-related key genes, and a diagnostic model was constructed. The ROC curve demonstrated that the model exhibited excellent diagnostic performance. DDX58, IFIT2, IFIH1, RSAD2, IFI44L, OAS2, OAS1, OASL, IFIT1, IFIT3, MX1, STAT1, and ISG15 were hub genes in TB, and the diagnostic model based on eight immune-related key genes exhibited good diagnostic performance.
期刊介绍:
AMB Express is a high quality journal that brings together research in the area of Applied and Industrial Microbiology with a particular interest in ''White Biotechnology'' and ''Red Biotechnology''. The emphasis is on processes employing microorganisms, eukaryotic cell cultures or enzymes for the biosynthesis, transformation and degradation of compounds. This includes fine and bulk chemicals, polymeric compounds and enzymes or other proteins. Downstream processes are also considered. Integrated processes combining biochemical and chemical processes are also published.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
