Jia Wang, Zelin Zhao, Min Guo, Liang Xiao, Haolin Tang, Jiantao Li, Zongkui Kou and Junsheng Li
{"title":"Epitaxial heterointerfacial electron bridge synchronizes oxygen evolution activity and stability on a layered double hydroxide surface†","authors":"Jia Wang, Zelin Zhao, Min Guo, Liang Xiao, Haolin Tang, Jiantao Li, Zongkui Kou and Junsheng Li","doi":"10.1039/D4EY00037D","DOIUrl":null,"url":null,"abstract":"<p >Scalable green hydrogen production <em>via</em> electrocatalytic water splitting is largely restricted by the insufficient activity and stability of oxygen evolution reaction (OER) catalysts at the anode. As a class of the most active OER catalysts in alkaline electrolyzers, the application of layered double hydroxides (LDHs) remains a main challenge owing to the unstable lattice oxygen dissolution due to the dominant lattice oxygen-involving OER mechanism during long-term operation. Herein, we found that using an epitaxial hetero-interfacing nickel hydroxide (namely Ni(OH)<small><sub>2</sub></small>) as an electron bridge between an active FeCo LDH and Ni foam support to form an LDH*/NFO catalyst, the electronic storage capacity around the Fermi level (−0.5 to +0.5 eV, e-D<small><sub>FE</sub></small>) sharply increases from 0.93 per cell to 1.51 per cell. Subsequently, we demonstrate that this high e-D<small><sub>FE</sub></small> enables ceaseless and fast power injection into the kinetic process of intermediate species conversion and inhibits lattice oxygen dissolution in the active FeCo LDH. Consequently, it demonstrated a low OER overpotential of 246 mV at a current density of 100 mA cm<small><sup>−2</sup></small> and ultrahigh stability for up to 3500 hours with an ultraslow overpotential increase rate of 9.4 × 10<small><sup>−3</sup></small> mV h<small><sup>−1</sup></small>. Therefore, we developed an epitaxial hetero-interfacial electron bridging strategy to synchronize the activity and stability of available catalysts for scalable green hydrogen production <em>via</em> electrocatalytic water splitting.</p>","PeriodicalId":72877,"journal":{"name":"EES catalysis","volume":" 3","pages":" 862-873"},"PeriodicalIF":0.0000,"publicationDate":"2024-04-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/ey/d4ey00037d?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EES catalysis","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ey/d4ey00037d","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
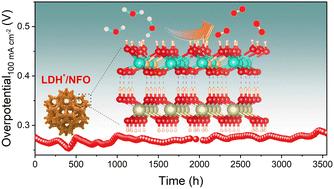
Scalable green hydrogen production via electrocatalytic water splitting is largely restricted by the insufficient activity and stability of oxygen evolution reaction (OER) catalysts at the anode. As a class of the most active OER catalysts in alkaline electrolyzers, the application of layered double hydroxides (LDHs) remains a main challenge owing to the unstable lattice oxygen dissolution due to the dominant lattice oxygen-involving OER mechanism during long-term operation. Herein, we found that using an epitaxial hetero-interfacing nickel hydroxide (namely Ni(OH)2) as an electron bridge between an active FeCo LDH and Ni foam support to form an LDH*/NFO catalyst, the electronic storage capacity around the Fermi level (−0.5 to +0.5 eV, e-DFE) sharply increases from 0.93 per cell to 1.51 per cell. Subsequently, we demonstrate that this high e-DFE enables ceaseless and fast power injection into the kinetic process of intermediate species conversion and inhibits lattice oxygen dissolution in the active FeCo LDH. Consequently, it demonstrated a low OER overpotential of 246 mV at a current density of 100 mA cm−2 and ultrahigh stability for up to 3500 hours with an ultraslow overpotential increase rate of 9.4 × 10−3 mV h−1. Therefore, we developed an epitaxial hetero-interfacial electron bridging strategy to synchronize the activity and stability of available catalysts for scalable green hydrogen production via electrocatalytic water splitting.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
