{"title":"RARS1‐related hypomyelinating leukodystrophy‐9 (HLD‐9) in two distinct Iranian families: Case report and literature review","authors":"Sajjad Biglari, Hassan Vahidnezhad, Mohammad Amin Tabatabaiefar, Hamid Reza Khorram Khorshid, Emran Esmaeilzadeh","doi":"10.1002/mgg3.2435","DOIUrl":null,"url":null,"abstract":"BackgroundHypomyelinating leukodystrophy‐9 (HLD‐9) is caused by biallelic pathogenic variants in <jats:italic>RARS1</jats:italic>, which codes for the cytoplasmic tRNA synthetase for arginine (ArgRS). This study aims to evaluate the clinical, neuroradiological, and genetic characteristics of patients with RARS1‐related disease and determine probable genotype–phenotype relationships.MethodsWe identified three patients with <jats:italic>RARS1</jats:italic> homozygous pathogenic variants. Furthermore, we performed a comprehensive review of the literature.ResultsHomozygous variants of <jats:italic>RARS1</jats:italic> (c.2T>C (p.Met1Thr)) were identified in three patients with HLD‐9. Clinical symptoms were severe in all patients. Following the literature review, thirty HLD‐9 cases from eight studies were found. The 33 patients' main symptoms were hypomyelination, language delay, and intellectual disability or developmental delay. The mean age of onset for HLD9 in the group of 33 patients with a known age of onset was 5.8 months (SD = 8.1). The interquartile range of age of onset was 0–10 months. Of the 25 variants identified, c.5A>G (p.Asp2Gly) was identified in 11 patients.ConclusionPathogenic variants in <jats:italic>RARS1</jats:italic> decrease ArgRS activity and cause a wide range of symptoms, from severe, early onset epileptic encephalopathy with brain atrophy to a mild condition with relatively maintained myelination. These symptoms include the classic hypomyelination presentation with nystagmus and spasticity. Furthermore, the pathogenicity of the variation c.2T>C (p.Met1Thr) has been shown.","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":null,"pages":null},"PeriodicalIF":1.5000,"publicationDate":"2024-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.2435","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
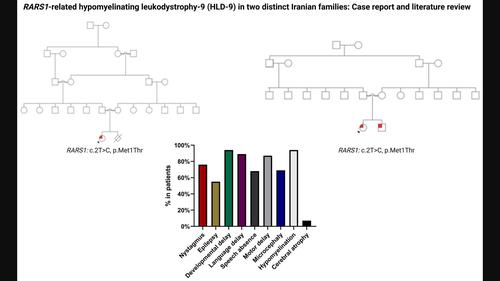
BackgroundHypomyelinating leukodystrophy‐9 (HLD‐9) is caused by biallelic pathogenic variants in RARS1, which codes for the cytoplasmic tRNA synthetase for arginine (ArgRS). This study aims to evaluate the clinical, neuroradiological, and genetic characteristics of patients with RARS1‐related disease and determine probable genotype–phenotype relationships.MethodsWe identified three patients with RARS1 homozygous pathogenic variants. Furthermore, we performed a comprehensive review of the literature.ResultsHomozygous variants of RARS1 (c.2T>C (p.Met1Thr)) were identified in three patients with HLD‐9. Clinical symptoms were severe in all patients. Following the literature review, thirty HLD‐9 cases from eight studies were found. The 33 patients' main symptoms were hypomyelination, language delay, and intellectual disability or developmental delay. The mean age of onset for HLD9 in the group of 33 patients with a known age of onset was 5.8 months (SD = 8.1). The interquartile range of age of onset was 0–10 months. Of the 25 variants identified, c.5A>G (p.Asp2Gly) was identified in 11 patients.ConclusionPathogenic variants in RARS1 decrease ArgRS activity and cause a wide range of symptoms, from severe, early onset epileptic encephalopathy with brain atrophy to a mild condition with relatively maintained myelination. These symptoms include the classic hypomyelination presentation with nystagmus and spasticity. Furthermore, the pathogenicity of the variation c.2T>C (p.Met1Thr) has been shown.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
