{"title":"SeqKit2: A Swiss army knife for sequence and alignment processing","authors":"Wei Shen, Botond Sipos, Liuyang Zhao","doi":"10.1002/imt2.191","DOIUrl":null,"url":null,"abstract":"<p>In the era of ubiquitous high-throughput sequencing studies, there is a growing need for analysis tools that are not just performant but also comprehensive and user-friendly enough to cater to both novice and advanced users. This article introduces SeqKit2, the next iteration of the widely used sequence analysis tool SeqKit, featuring expanded functionality, performance optimizations, and support for additional compression methods. Retaining a pragmatic subcommand architecture, SeqKit2 represents substantial enhancement through the inclusion of 19 additional subcommands, expanding its overall repertoire to a total of 38 in eight categories. The new subcommands add functionality such as amplicon processing and robust, error-tolerant parsing of sequence records. In addition, three subcommands designed for real-time analysis are added for periodic monitoring of properties of FASTQ and Binary Alignment/Map alignment records and real-time streaming from multiple sequence files. The performance of SeqKit2 is benchmarked against the old version of SeqKit, Bioawk, Seqtk, and SeqFu tools. SeqKit2 consistently outperforms its predecessor, albeit with marginally higher memory usage, while maintaining competitive runtimes against other tools. With its broad functionality, proven usability, and ongoing development driven by user feedback, we hope that bioinformaticians will find SeqKit2 useful as a “Swiss army knife” of sequence and alignment processing—equally adept at facilitating ad hoc analyses and seamlessly integrating into larger pipelines.</p>","PeriodicalId":73342,"journal":{"name":"iMeta","volume":"3 3","pages":""},"PeriodicalIF":23.7000,"publicationDate":"2024-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/imt2.191","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"iMeta","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/imt2.191","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
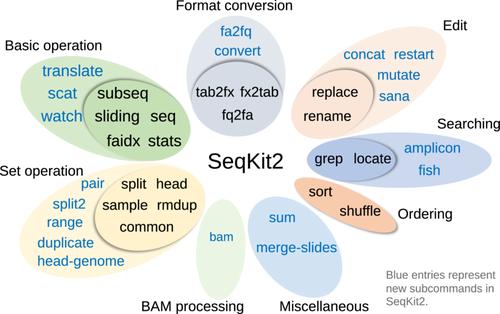
In the era of ubiquitous high-throughput sequencing studies, there is a growing need for analysis tools that are not just performant but also comprehensive and user-friendly enough to cater to both novice and advanced users. This article introduces SeqKit2, the next iteration of the widely used sequence analysis tool SeqKit, featuring expanded functionality, performance optimizations, and support for additional compression methods. Retaining a pragmatic subcommand architecture, SeqKit2 represents substantial enhancement through the inclusion of 19 additional subcommands, expanding its overall repertoire to a total of 38 in eight categories. The new subcommands add functionality such as amplicon processing and robust, error-tolerant parsing of sequence records. In addition, three subcommands designed for real-time analysis are added for periodic monitoring of properties of FASTQ and Binary Alignment/Map alignment records and real-time streaming from multiple sequence files. The performance of SeqKit2 is benchmarked against the old version of SeqKit, Bioawk, Seqtk, and SeqFu tools. SeqKit2 consistently outperforms its predecessor, albeit with marginally higher memory usage, while maintaining competitive runtimes against other tools. With its broad functionality, proven usability, and ongoing development driven by user feedback, we hope that bioinformaticians will find SeqKit2 useful as a “Swiss army knife” of sequence and alignment processing—equally adept at facilitating ad hoc analyses and seamlessly integrating into larger pipelines.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
