Xi Peng, Kai Feng, Xingsheng Yang, Qing He, Bo Zhao, Tong Li, Shang Wang, Ye Deng
{"title":"iNAP 2.0: Harnessing metabolic complementarity in microbial network analysis","authors":"Xi Peng, Kai Feng, Xingsheng Yang, Qing He, Bo Zhao, Tong Li, Shang Wang, Ye Deng","doi":"10.1002/imt2.235","DOIUrl":null,"url":null,"abstract":"<p>With the widespread adoption of metagenomic sequencing, new perspectives have emerged for studying microbial ecological networks, yielding metabolic evidence of interspecies interactions that traditional co-occurrence networks cannot infer. This protocol introduces the integrated Network Analysis Pipeline 2.0 (iNAP 2.0), which features an innovative metabolic complementarity network for microbial studies from metagenomics sequencing data. iNAP 2.0 sets up a four-module process for metabolic interaction analysis, namely: (I) Prepare genome-scale metabolic models; (II) Infer pairwise interactions of genome-scale metabolic models; (III) Construct metabolic interaction networks; and (IV) Analyze metabolic interaction networks. Starting from metagenome-assembled or complete genomes, iNAP 2.0 offers a variety of methods to quantify the potential and trends of metabolic complementarity between models, including the PhyloMint pipeline based on phylogenetic distance-adjusted metabolic complementarity, the SMETANA (species metabolic interaction analysis) approach based on cross-feeding substrate exchange prediction, and metabolic distance calculation based on parsimonious flux balance analysis (pFBA). Notably, iNAP 2.0 integrates the random matrix theory (RMT) approach to find the suitable threshold for metabolic interaction network construction. Finally, the metabolic interaction networks can proceed to analysis using topological feature analysis such as hub node determination. In addition, a key feature of iNAP 2.0 is the identification of potentially transferable metabolites between species, presented as intermediate nodes that connect microbial nodes in the metabolic complementarity network. To illustrate these new features, we use a set of metagenome-assembled genomes as an example to comprehensively document the usage of the tools. iNAP 2.0 is available at https://inap.denglab.org.cn for all users to register and use for free.</p>","PeriodicalId":73342,"journal":{"name":"iMeta","volume":"3 5","pages":""},"PeriodicalIF":23.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/imt2.235","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"iMeta","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/imt2.235","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
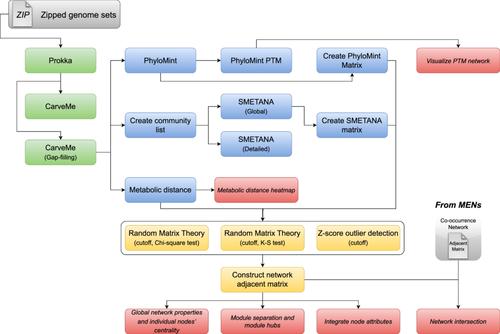
With the widespread adoption of metagenomic sequencing, new perspectives have emerged for studying microbial ecological networks, yielding metabolic evidence of interspecies interactions that traditional co-occurrence networks cannot infer. This protocol introduces the integrated Network Analysis Pipeline 2.0 (iNAP 2.0), which features an innovative metabolic complementarity network for microbial studies from metagenomics sequencing data. iNAP 2.0 sets up a four-module process for metabolic interaction analysis, namely: (I) Prepare genome-scale metabolic models; (II) Infer pairwise interactions of genome-scale metabolic models; (III) Construct metabolic interaction networks; and (IV) Analyze metabolic interaction networks. Starting from metagenome-assembled or complete genomes, iNAP 2.0 offers a variety of methods to quantify the potential and trends of metabolic complementarity between models, including the PhyloMint pipeline based on phylogenetic distance-adjusted metabolic complementarity, the SMETANA (species metabolic interaction analysis) approach based on cross-feeding substrate exchange prediction, and metabolic distance calculation based on parsimonious flux balance analysis (pFBA). Notably, iNAP 2.0 integrates the random matrix theory (RMT) approach to find the suitable threshold for metabolic interaction network construction. Finally, the metabolic interaction networks can proceed to analysis using topological feature analysis such as hub node determination. In addition, a key feature of iNAP 2.0 is the identification of potentially transferable metabolites between species, presented as intermediate nodes that connect microbial nodes in the metabolic complementarity network. To illustrate these new features, we use a set of metagenome-assembled genomes as an example to comprehensively document the usage of the tools. iNAP 2.0 is available at https://inap.denglab.org.cn for all users to register and use for free.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
