Dimitrios Petsopoulos, Jordan P. Cuff, James R. Bell, James J. N. Kitson, Larissa Collins, Neil Boonham, Ramiro Morales-Hojas, Darren M. Evans
{"title":"Identifying archived insect bulk samples using DNA metabarcoding: A case study using the long-term Rothamsted Insect Survey","authors":"Dimitrios Petsopoulos, Jordan P. Cuff, James R. Bell, James J. N. Kitson, Larissa Collins, Neil Boonham, Ramiro Morales-Hojas, Darren M. Evans","doi":"10.1002/edn3.542","DOIUrl":null,"url":null,"abstract":"<p>Insect populations are declining in many parts of the world, but a lack of long-term monitoring data is impeding our ability to understand and mitigate the causes of insect biodiversity loss. Whilst high-throughput sequencing (HTS) approaches, such as DNA metabarcoding, have the potential to revolutionize insect biomonitoring through rapid scalable identification, it is unclear to what extent HTS can be applied to long-term stored insect samples. Archived insect samples could inform forecasting and provide valuable information regarding past changes to biodiversity. Here, we assess the efficacy of DNA metabarcoding to identify archived samples from the longest passive monitoring scheme in the United Kingdom: the Rothamsted Insect Survey (RIS). With a focus on aphids as the target taxa of a national network of suction-traps, we analyze a 16-year time-series of stored samples (2003–2018) using DNA metabarcoding from one of the RIS suction traps as an exemplar. We achieved this by using a non-destructive DNA extraction protocol, ensuring the integrity of archival samples for further studies. We compared the identities of aphids determined by both metabarcoding (as inferred amplicon sequence variants [ASVs]) and morphological identification and found that metabarcoding detected most genera with varying success (mean > 76%). When comparing the two methods objectively (i.e., including taxa not detected morphologically), however, congruence decreased (51%). We show that minimum sequence copy thresholds can minimize metabarcoding false positives, but at the expense of introducing false negatives, highlighting the need for careful data curation. Detectability of taxa identified morphologically and similarity between the two methods did not significantly vary over time, demonstrating the viability of metabarcoding for screening archival samples. We discuss the advantages and challenges of metabarcoding for insect biomonitoring, particularly from archival samples, including improvements to sample handling, processing, and archiving. We highlight the wider potential of HTS approaches for stored samples from insect monitoring schemes, unlocking the immense potential of global historical time series.</p>","PeriodicalId":52828,"journal":{"name":"Environmental DNA","volume":"6 3","pages":""},"PeriodicalIF":6.2000,"publicationDate":"2024-05-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/edn3.542","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental DNA","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/edn3.542","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
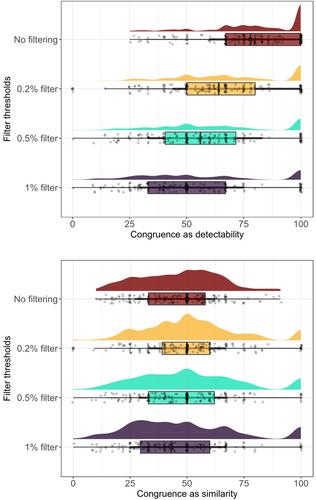
Insect populations are declining in many parts of the world, but a lack of long-term monitoring data is impeding our ability to understand and mitigate the causes of insect biodiversity loss. Whilst high-throughput sequencing (HTS) approaches, such as DNA metabarcoding, have the potential to revolutionize insect biomonitoring through rapid scalable identification, it is unclear to what extent HTS can be applied to long-term stored insect samples. Archived insect samples could inform forecasting and provide valuable information regarding past changes to biodiversity. Here, we assess the efficacy of DNA metabarcoding to identify archived samples from the longest passive monitoring scheme in the United Kingdom: the Rothamsted Insect Survey (RIS). With a focus on aphids as the target taxa of a national network of suction-traps, we analyze a 16-year time-series of stored samples (2003–2018) using DNA metabarcoding from one of the RIS suction traps as an exemplar. We achieved this by using a non-destructive DNA extraction protocol, ensuring the integrity of archival samples for further studies. We compared the identities of aphids determined by both metabarcoding (as inferred amplicon sequence variants [ASVs]) and morphological identification and found that metabarcoding detected most genera with varying success (mean > 76%). When comparing the two methods objectively (i.e., including taxa not detected morphologically), however, congruence decreased (51%). We show that minimum sequence copy thresholds can minimize metabarcoding false positives, but at the expense of introducing false negatives, highlighting the need for careful data curation. Detectability of taxa identified morphologically and similarity between the two methods did not significantly vary over time, demonstrating the viability of metabarcoding for screening archival samples. We discuss the advantages and challenges of metabarcoding for insect biomonitoring, particularly from archival samples, including improvements to sample handling, processing, and archiving. We highlight the wider potential of HTS approaches for stored samples from insect monitoring schemes, unlocking the immense potential of global historical time series.
利用 DNA 代谢编码鉴定归档的昆虫大宗样本:利用长期的 Rothamsted 昆虫调查进行案例研究
世界上许多地方的昆虫数量都在减少,但长期监测数据的缺乏阻碍了我们了解和减轻昆虫生物多样性丧失原因的能力。虽然高通量测序(HTS)方法(如 DNA 代谢编码)有可能通过快速可扩展的鉴定彻底改变昆虫生物监测工作,但目前还不清楚 HTS 在多大程度上可应用于长期储存的昆虫样本。存档昆虫样本可以为预测提供信息,并提供有关生物多样性过去变化的宝贵信息。在此,我们评估了 DNA 代谢编码在鉴定英国最长的被动监测计划--Rothamsted 昆虫调查(RIS)中的存档样本方面的功效。蚜虫是吸式捕集器全国网络的目标分类群,我们以蚜虫捕集器吸式捕集器为范例,利用 DNA 元标定技术分析了 16 年(2003-2018 年)的存储样本时间序列。我们采用非破坏性的 DNA 提取方案实现了这一目标,确保了进一步研究中档案样本的完整性。我们比较了通过元标码(推断出的扩增子序列变异[ASV])和形态鉴定两种方法确定的蚜虫身份,发现元标码能检测出大多数属,但成功率不一(平均为 76%)。然而,当客观地比较这两种方法(即包括形态学上未检测到的类群)时,一致性下降(51%)。我们的研究表明,最小序列拷贝阈值可以最大限度地减少元标码的假阳性,但其代价是引入假阴性,这就强调了对数据进行仔细整理的必要性。通过形态学鉴定的类群的可检测性和两种方法之间的相似性并没有随着时间的推移而发生显著变化,这证明了元条码在筛选档案样本方面的可行性。我们讨论了元条码在昆虫生物监测方面的优势和挑战,特别是来自档案样本的优势和挑战,包括样本处理、加工和存档方面的改进。我们强调了 HTS 方法在昆虫监测计划的存储样本方面的更广泛潜力,从而释放全球历史时间序列的巨大潜力。



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
