Jun Hu , Zhe Li , Bing Rao , Maha A. Thafar , Muhammad Arif
{"title":"Improving protein-protein interaction prediction using protein language model and protein network features","authors":"Jun Hu , Zhe Li , Bing Rao , Maha A. Thafar , Muhammad Arif","doi":"10.1016/j.ab.2024.115550","DOIUrl":null,"url":null,"abstract":"<div><p>Interactions between proteins are ubiquitous in a wide variety of biological processes. Accurately identifying the protein-protein interaction (PPI) is of significant importance for understanding the mechanisms of protein functions and facilitating drug discovery. Although the wet-lab technological methods are the best way to identify PPI, their major constraints are their time-consuming nature, high cost, and labor-intensiveness. Hence, lots of efforts have been made towards developing computational methods to improve the performance of PPI prediction. In this study, we propose a novel hybrid computational method (called KSGPPI) that aims at improving the prediction performance of PPI via extracting the discriminative information from protein sequences and interaction networks. The KSGPPI model comprises two feature extraction modules. In the first feature extraction module, a large protein language model, ESM-2, is employed to exploit the global complex patterns concealed within protein sequences. Subsequently, feature representations are further extracted through CKSAAP, and a two-dimensional convolutional neural network (CNN) is utilized to capture local information. In the second feature extraction module, the query protein acquires its similar protein from the STRING database via the sequence alignment tool NW-align and then captures the graph embedding feature for the query protein in the protein interaction network of the similar protein using the algorithm of Node2vec. Finally, the features of these two feature extraction modules are efficiently fused; the fused features are then fed into the multilayer perceptron to predict PPI. The results of five-fold cross-validation on the used benchmarked datasets demonstrate that KSGPPI achieves an average prediction accuracy of 88.96 %. Additionally, the average Matthews correlation coefficient value (0.781) of KSGPPI is significantly higher than that of those state-of-the-art PPI prediction methods. The standalone package of KSGPPI is freely downloaded at <span>https://github.com/rickleezhe/KSGPPI</span><svg><path></path></svg>.</p></div>","PeriodicalId":7830,"journal":{"name":"Analytical biochemistry","volume":"693 ","pages":"Article 115550"},"PeriodicalIF":2.5000,"publicationDate":"2024-04-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Analytical biochemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0003269724000940","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
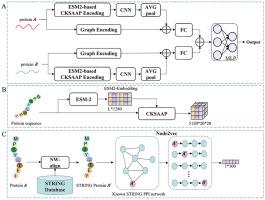
Interactions between proteins are ubiquitous in a wide variety of biological processes. Accurately identifying the protein-protein interaction (PPI) is of significant importance for understanding the mechanisms of protein functions and facilitating drug discovery. Although the wet-lab technological methods are the best way to identify PPI, their major constraints are their time-consuming nature, high cost, and labor-intensiveness. Hence, lots of efforts have been made towards developing computational methods to improve the performance of PPI prediction. In this study, we propose a novel hybrid computational method (called KSGPPI) that aims at improving the prediction performance of PPI via extracting the discriminative information from protein sequences and interaction networks. The KSGPPI model comprises two feature extraction modules. In the first feature extraction module, a large protein language model, ESM-2, is employed to exploit the global complex patterns concealed within protein sequences. Subsequently, feature representations are further extracted through CKSAAP, and a two-dimensional convolutional neural network (CNN) is utilized to capture local information. In the second feature extraction module, the query protein acquires its similar protein from the STRING database via the sequence alignment tool NW-align and then captures the graph embedding feature for the query protein in the protein interaction network of the similar protein using the algorithm of Node2vec. Finally, the features of these two feature extraction modules are efficiently fused; the fused features are then fed into the multilayer perceptron to predict PPI. The results of five-fold cross-validation on the used benchmarked datasets demonstrate that KSGPPI achieves an average prediction accuracy of 88.96 %. Additionally, the average Matthews correlation coefficient value (0.781) of KSGPPI is significantly higher than that of those state-of-the-art PPI prediction methods. The standalone package of KSGPPI is freely downloaded at https://github.com/rickleezhe/KSGPPI.
期刊介绍:
The journal''s title Analytical Biochemistry: Methods in the Biological Sciences declares its broad scope: methods for the basic biological sciences that include biochemistry, molecular genetics, cell biology, proteomics, immunology, bioinformatics and wherever the frontiers of research take the field.
The emphasis is on methods from the strictly analytical to the more preparative that would include novel approaches to protein purification as well as improvements in cell and organ culture. The actual techniques are equally inclusive ranging from aptamers to zymology.
The journal has been particularly active in:
-Analytical techniques for biological molecules-
Aptamer selection and utilization-
Biosensors-
Chromatography-
Cloning, sequencing and mutagenesis-
Electrochemical methods-
Electrophoresis-
Enzyme characterization methods-
Immunological approaches-
Mass spectrometry of proteins and nucleic acids-
Metabolomics-
Nano level techniques-
Optical spectroscopy in all its forms.
The journal is reluctant to include most drug and strictly clinical studies as there are more suitable publication platforms for these types of papers.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
