{"title":"Computational assessment of the use of graphene-based nanosheets as PtII chemotherapeutics delivery systems","authors":"Daniele Belletto, Vincenzo Vigna, Pierraffaele Barretta, Fortuna Ponte, Gloria Mazzone, Stefano Scoditti, Emilia Sicilia","doi":"10.1002/jcc.27394","DOIUrl":null,"url":null,"abstract":"<p>Graphene is the newest form of elemental carbon and it is becoming rapidly a potential candidate in the framework of nano-bio research. Many reports confirm the successful use of graphene-based materials as carriers of anticancer drugs having relatively high loading capacities compared with other nanocarriers. Here, the outcomes of a systematic study of the adsorption behavior of FDA approved Pt<sup>II</sup> drugs cisplatin, oxaliplatin, and carboplatin on surface models of pristine, holey, and nitrogen-doped holey graphene are reported. DFT investigations in water solvent have been carried out considering several initial orientations of the drugs with respect to the surfaces. Adsorption free energies, calculated including basis set superposition error (BSSE) corrections, result to be significantly negative for many of the drug@carrier adducts indicating that tested layers could be used as potential carriers for the delivery of anticancer Pt<sup>II</sup> drugs. The reduced density gradient (RDG) analysis allows to show that many kinds of non-covalent interactions, including canonical H-bond, are responsible for the stabilization of the formed adducts.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 24","pages":"2059-2070"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27394","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
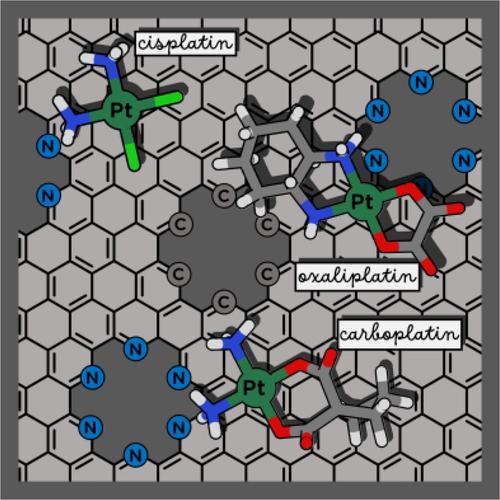
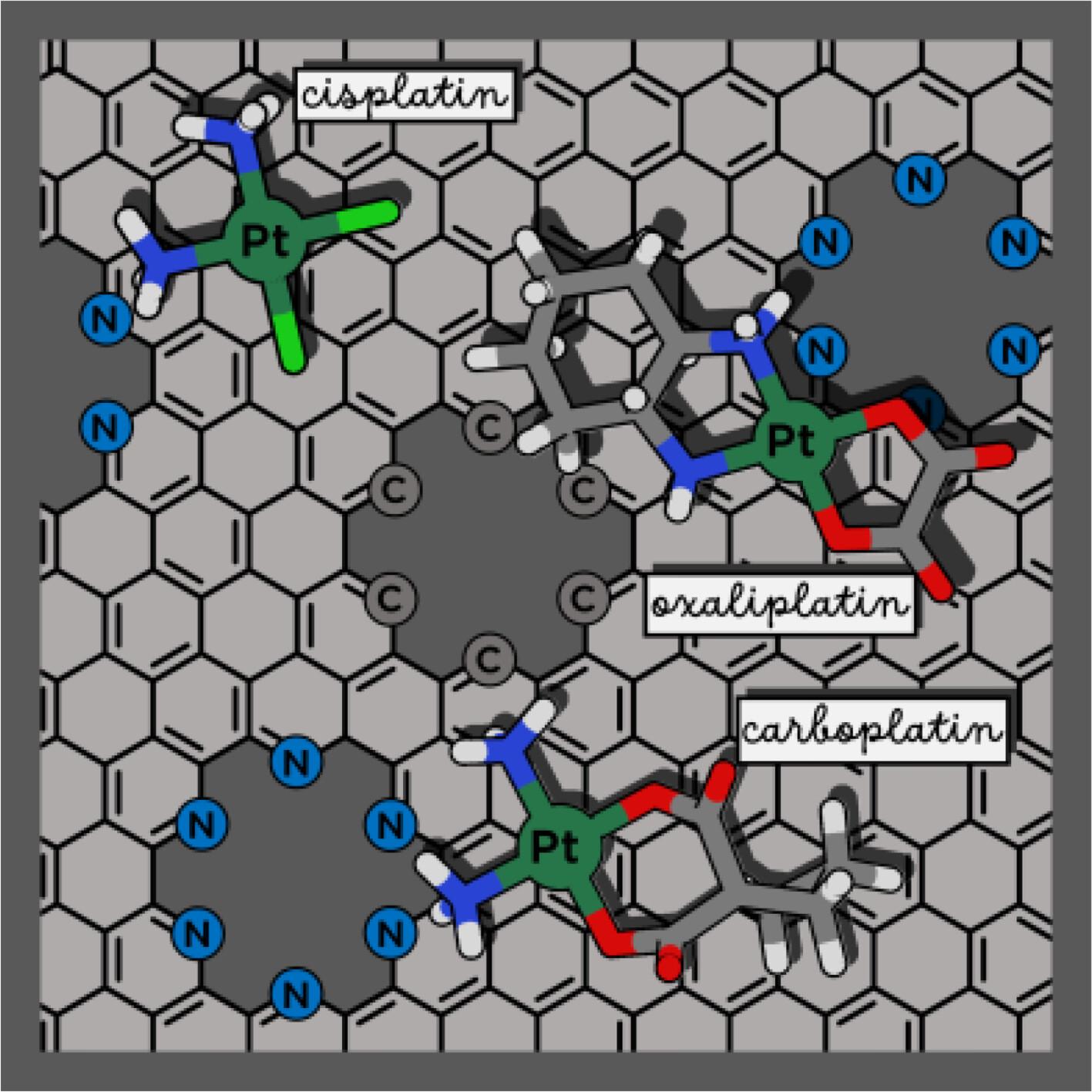
Graphene is the newest form of elemental carbon and it is becoming rapidly a potential candidate in the framework of nano-bio research. Many reports confirm the successful use of graphene-based materials as carriers of anticancer drugs having relatively high loading capacities compared with other nanocarriers. Here, the outcomes of a systematic study of the adsorption behavior of FDA approved PtII drugs cisplatin, oxaliplatin, and carboplatin on surface models of pristine, holey, and nitrogen-doped holey graphene are reported. DFT investigations in water solvent have been carried out considering several initial orientations of the drugs with respect to the surfaces. Adsorption free energies, calculated including basis set superposition error (BSSE) corrections, result to be significantly negative for many of the drug@carrier adducts indicating that tested layers could be used as potential carriers for the delivery of anticancer PtII drugs. The reduced density gradient (RDG) analysis allows to show that many kinds of non-covalent interactions, including canonical H-bond, are responsible for the stabilization of the formed adducts.
石墨烯是元素碳的最新形式,正迅速成为纳米生物研究领域的潜在候选材料。许多报道证实,石墨烯基材料可成功用作抗癌药物的载体,与其他纳米载体相比,它具有相对较高的负载能力。本文报告了对美国 FDA 批准的铂贰类药物顺铂、奥沙利铂和卡铂在原始石墨烯、空洞石墨烯和掺氮空洞石墨烯表面模型上的吸附行为进行系统研究的结果。考虑到药物相对于表面的几种初始取向,在水溶剂中进行了 DFT 研究。计算得出的吸附自由能(包括基集叠加误差 (BSSE) 修正)对许多药物@载体加合物来说都是显著的负值,这表明测试的石墨烯层可用作输送抗癌铂铱药物的潜在载体。还原密度梯度(RDG)分析表明,包括典型 H 键在内的多种非共价相互作用是所形成加合物稳定的原因。
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
