Holly Mabillard, Rebecca Ryan, Nik Tzoumas, Susie Gear, John A Sayer
{"title":"Explaining Alport syndrome-lessons from the adult nephrology clinic.","authors":"Holly Mabillard, Rebecca Ryan, Nik Tzoumas, Susie Gear, John A Sayer","doi":"10.1007/s44162-024-00036-z","DOIUrl":null,"url":null,"abstract":"<p><p>Alport syndrome is a genetic kidney disease that causes worsening of kidney function over time, often progressing to kidney failure. Some types of Alport syndrome cause other symptoms and signs, including hearing loss and eye abnormalities. Research now indicates that Alport syndrome (autosomal dominant inheritance) is the most common form. Alport syndrome can have X-linked or a rare form of autosomal recessive inheritance. Traditionally, a kidney biopsy was used to diagnose Alport syndrome, but genetic testing provides a more precise and less invasive means of diagnosis and reveals the underlying pattern of inheritance. At present, there are no specific curative treatments for Alport syndrome however there is a strong international effort in pursuit of future therapies. Currently, angiotensin-converting enzyme inhibitors (ACEi), or an angiotensin receptor blocker (ARB) if a patient cannot tolerate an ACEi, slow down the progression of kidney disease and can delay the onset of kidney failure by years. There are other potential treatments in research that potentially can help delay the onset of kidney issues. Early treatment of patients and identification of their at-risk relatives is a priority. People living with Alport syndrome and their doctors now benefit from an active international research community working on translating further treatments into clinical practice and providing up-to-date clinical guidelines.</p>","PeriodicalId":73925,"journal":{"name":"Journal of rare diseases (Berlin, Germany)","volume":"3 1","pages":"14"},"PeriodicalIF":0.0000,"publicationDate":"2024-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11088994/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of rare diseases (Berlin, Germany)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s44162-024-00036-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/13 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
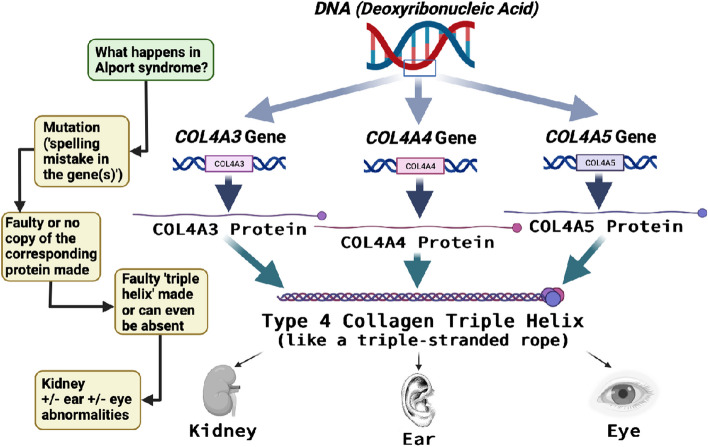
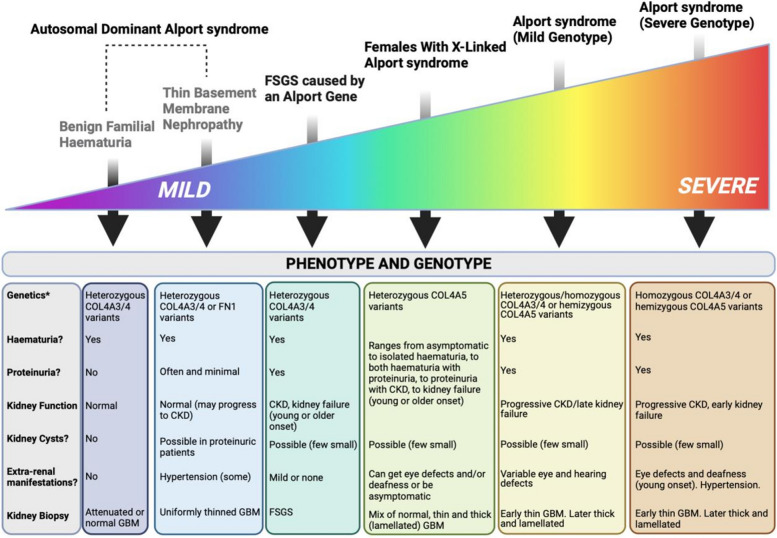
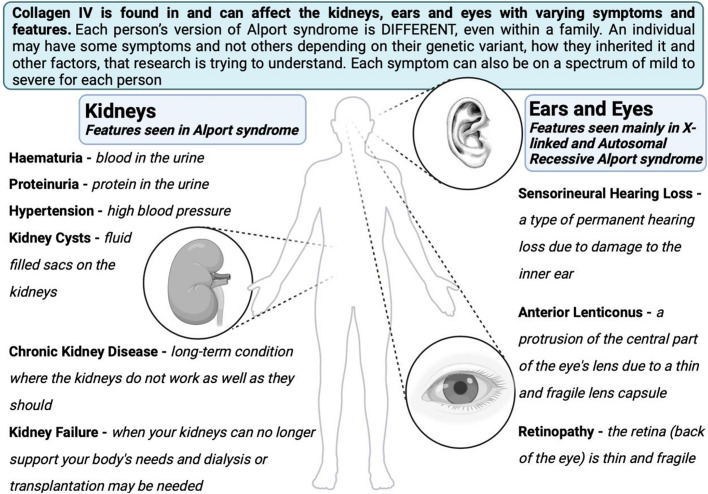
Alport syndrome is a genetic kidney disease that causes worsening of kidney function over time, often progressing to kidney failure. Some types of Alport syndrome cause other symptoms and signs, including hearing loss and eye abnormalities. Research now indicates that Alport syndrome (autosomal dominant inheritance) is the most common form. Alport syndrome can have X-linked or a rare form of autosomal recessive inheritance. Traditionally, a kidney biopsy was used to diagnose Alport syndrome, but genetic testing provides a more precise and less invasive means of diagnosis and reveals the underlying pattern of inheritance. At present, there are no specific curative treatments for Alport syndrome however there is a strong international effort in pursuit of future therapies. Currently, angiotensin-converting enzyme inhibitors (ACEi), or an angiotensin receptor blocker (ARB) if a patient cannot tolerate an ACEi, slow down the progression of kidney disease and can delay the onset of kidney failure by years. There are other potential treatments in research that potentially can help delay the onset of kidney issues. Early treatment of patients and identification of their at-risk relatives is a priority. People living with Alport syndrome and their doctors now benefit from an active international research community working on translating further treatments into clinical practice and providing up-to-date clinical guidelines.
阿尔波特综合征是一种遗传性肾脏疾病,会随着时间的推移导致肾功能恶化,通常会发展为肾衰竭。某些类型的阿尔波特综合征还会引起其他症状和体征,包括听力损失和眼睛异常。目前的研究表明,阿尔波特综合征(常染色体显性遗传)是最常见的类型。阿尔波特综合征可以是 X 连锁遗传,也可以是罕见的常染色体隐性遗传。传统上,肾活检被用来诊断阿尔波特综合征,但基因检测提供了一种更精确、创伤更小的诊断手段,并能揭示潜在的遗传模式。目前,阿尔波特综合征还没有特效的治疗方法,但国际上正在大力寻求未来的治疗方法。目前,血管紧张素转换酶抑制剂(ACEi)或血管紧张素受体阻滞剂(ARB)(如果患者不能耐受血管紧张素转换酶抑制剂)可减缓肾病的进展,并可将肾衰竭的发病时间推迟数年。研究中还有其他潜在的治疗方法,可能有助于延缓肾脏问题的发生。患者的早期治疗和高危亲属的识别是当务之急。现在,阿尔波特综合征患者和他们的医生都受益于一个活跃的国际研究团体,该团体致力于将进一步的治疗方法转化为临床实践,并提供最新的临床指南。


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
