{"title":"Mapping the correlations between bandgap, HOMO, and LUMO trends for meta substituted Zn-MOFs","authors":"Kyle I. Williamson, Daniel J. C. Herr, Yirong Mo","doi":"10.1002/jcc.27432","DOIUrl":null,"url":null,"abstract":"<p>Bandgap is a key property that determines electrical and optical properties in materials. Modulating the bandgap thus is critical in developing novel materials particularly semiconductors with improved features. This study examines the bandgap, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) energy level trends in a metal organic framework, metal–organic framework 5 (MOF-5), as a function of Hammett substituent effect (with the constant <i>σ</i><sub><i>m</i></sub> in the meta-position of the benzene ring) and solvent dielectric effect (with the constant <i>ε</i>). Specifically, experimental design and response surface methodologies helped to assess the significance of trends and correlations between these molecular properties with <i>σ</i><sub><i>m</i></sub> and <i>ε</i>. While the HOMO and LUMO decrease with increasing <i>σ</i><sub><i>m</i></sub>, the LUMO exhibits greater sensitivity to the substituent's electron withdrawing capability. The relative difference in these trends helps to explain why the bandgap tends to decrease with increasing <i>σ</i><sub><i>m</i></sub>.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 25","pages":"2119-2127"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27432","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27432","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
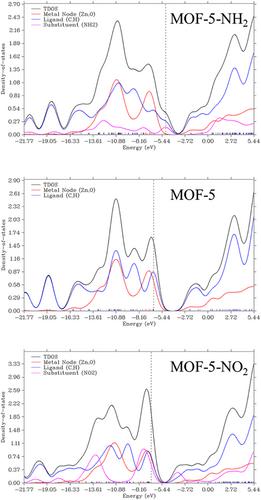
Bandgap is a key property that determines electrical and optical properties in materials. Modulating the bandgap thus is critical in developing novel materials particularly semiconductors with improved features. This study examines the bandgap, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) energy level trends in a metal organic framework, metal–organic framework 5 (MOF-5), as a function of Hammett substituent effect (with the constant σm in the meta-position of the benzene ring) and solvent dielectric effect (with the constant ε). Specifically, experimental design and response surface methodologies helped to assess the significance of trends and correlations between these molecular properties with σm and ε. While the HOMO and LUMO decrease with increasing σm, the LUMO exhibits greater sensitivity to the substituent's electron withdrawing capability. The relative difference in these trends helps to explain why the bandgap tends to decrease with increasing σm.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
