Christopher Vorreiter, Dina Robaa and Wolfgang Sippl*,
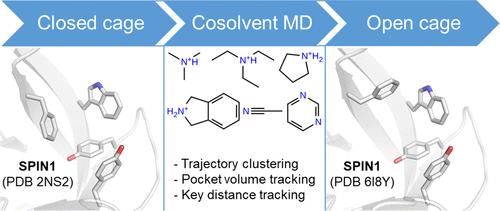
{"title":"Exploring Aromatic Cage Flexibility Using Cosolvent Molecular Dynamics Simulations─An In-Silico Case Study of Tudor Domains","authors":"Christopher Vorreiter, Dina Robaa and Wolfgang Sippl*, ","doi":"10.1021/acs.jcim.4c00298","DOIUrl":null,"url":null,"abstract":"<p >Cosolvent molecular dynamics (MD) simulations have proven to be powerful in silico tools to predict hotspots for binding regions on protein surfaces. In the current study, the method was adapted and applied to two Tudor domain-containing proteins, namely Spindlin1 (SPIN1) and survival motor neuron protein (SMN). Tudor domains are characterized by so-called aromatic cages that recognize methylated lysine residues of protein targets. In the study, the conformational transitions from closed to open aromatic cage conformations were investigated by performing MD simulations with cosolvents using six different probe molecules. It is shown that a trajectory clustering approach in combination with volume and atomic distance tracking allows a reasonable discrimination between open and closed aromatic cage conformations and the docking of inhibitors yields very good reproducibility with crystal structures. Cosolvent MDs are suitable to capture the flexibility of aromatic cages and thus represent a promising tool for the optimization of inhibitors.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"64 11","pages":"4553–4569"},"PeriodicalIF":5.3000,"publicationDate":"2024-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/epdf/10.1021/acs.jcim.4c00298","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c00298","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Cosolvent molecular dynamics (MD) simulations have proven to be powerful in silico tools to predict hotspots for binding regions on protein surfaces. In the current study, the method was adapted and applied to two Tudor domain-containing proteins, namely Spindlin1 (SPIN1) and survival motor neuron protein (SMN). Tudor domains are characterized by so-called aromatic cages that recognize methylated lysine residues of protein targets. In the study, the conformational transitions from closed to open aromatic cage conformations were investigated by performing MD simulations with cosolvents using six different probe molecules. It is shown that a trajectory clustering approach in combination with volume and atomic distance tracking allows a reasonable discrimination between open and closed aromatic cage conformations and the docking of inhibitors yields very good reproducibility with crystal structures. Cosolvent MDs are suitable to capture the flexibility of aromatic cages and thus represent a promising tool for the optimization of inhibitors.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
