Mechanistic study on C(sp2)–F bond activation by a Ni0(silyl)-ate complex: an outer-sphere pathway via Ni0-mediated nucleophilic aromatic substitution†
Xiao-Xia You , Jian-Sen Wang , Xiao-Xiao Li , Ling-Qi Meng , Rong-Lin Zhong , Zhong-Min Su
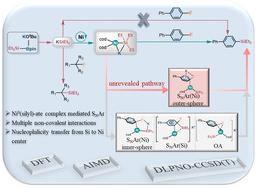
{"title":"Mechanistic study on C(sp2)–F bond activation by a Ni0(silyl)-ate complex: an outer-sphere pathway via Ni0-mediated nucleophilic aromatic substitution†","authors":"Xiao-Xia You , Jian-Sen Wang , Xiao-Xiao Li , Ling-Qi Meng , Rong-Lin Zhong , Zhong-Min Su","doi":"10.1039/d4qo00743c","DOIUrl":null,"url":null,"abstract":"<div><div>In this study, we conducted a theoretical investigation to clarify the mechanism of C–F bond activation using density functional theory (DFT) calculations and <em>ab initio</em> molecular dynamics (AIMD) simulations, with Et<sub>3</sub>SiBpin and KO<sup>t</sup>Bu serving as reaction partners. Results suggest that a reactive nucleophile KSiEt<sub>3</sub> is generated <em>in situ</em>, which activates the C(sp<sup>3</sup>)–F bond <em>via</em> a bimolecular nucleophilic substitution (S<sub>N</sub>2) with a lower energy barrier (23.6 kcal mol<sup>−1</sup>) than that (28.2 kcal mol<sup>−1</sup>) for activation of the C(sp<sup>2</sup>)–F bond <em>via</em> a nucleophilic aromatic substitution reaction (S<sub>N</sub>Ar). This is consistent with the experimental result that silylation of the C(sp<sup>3</sup>)–F bond succeeded without a Ni<sup>0</sup>-catalyst, while that of the C(sp<sup>2</sup>)–F bond did not. In the presence of Ni<sup>0</sup>(cod)<sub>2</sub>, a more reactive ate-complex K<sup>+</sup>[Ni<sup>0</sup>(cod)<sub>2</sub>(SiEt<sub>3</sub>)]<sup>−</sup> is generated <em>via</em> the strong coordination of the (SiEt<sub>3</sub>)<sup>−</sup> anion to the Ni<sup>0</sup>-center. The inert C(sp<sup>2</sup>)–F bond activation thus succeeded <em>via</em> an unusual Ni<sup>0</sup>-mediated S<sub>N</sub>Ar. Surprisingly, results show the activation energy (21.8 kcal mol<sup>−1</sup>) for nucleophilic attack by the Ni<sup>0</sup> center is significantly lower than that (36.2 kcal mol<sup>−1</sup>) by the (SiEt<sub>3</sub>)<sup>−</sup> anion. Furthermore, the outer-sphere transition state is more stable than the well-known inner-sphere one because the radius of K<sup>+</sup> is too large to form a multicomponent ring with synergistic stabilization. In addition to the Ni⋯Si coordination, this unprecedented transition state in the C(sp<sup>2</sup>)–F bond activation is stabilized through multiple non-covalent interactions. Additionally, AIMD simulations were employed to elucidate the dynamic effects of the Ni<sup>0</sup>(silyl)-ate complex initiated C(sp<sup>2</sup>)–F bond activation, highlighting the pivotal role of multiple non-covalent interactions in the reaction.</div></div>","PeriodicalId":94379,"journal":{"name":"Organic chemistry frontiers : an international journal of organic chemistry","volume":"11 14","pages":"Pages 3913-3923"},"PeriodicalIF":0.0000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organic chemistry frontiers : an international journal of organic chemistry","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/org/science/article/pii/S2052412924003905","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/5/21 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
In this study, we conducted a theoretical investigation to clarify the mechanism of C–F bond activation using density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations, with Et3SiBpin and KOtBu serving as reaction partners. Results suggest that a reactive nucleophile KSiEt3 is generated in situ, which activates the C(sp3)–F bond via a bimolecular nucleophilic substitution (SN2) with a lower energy barrier (23.6 kcal mol−1) than that (28.2 kcal mol−1) for activation of the C(sp2)–F bond via a nucleophilic aromatic substitution reaction (SNAr). This is consistent with the experimental result that silylation of the C(sp3)–F bond succeeded without a Ni0-catalyst, while that of the C(sp2)–F bond did not. In the presence of Ni0(cod)2, a more reactive ate-complex K+[Ni0(cod)2(SiEt3)]− is generated via the strong coordination of the (SiEt3)− anion to the Ni0-center. The inert C(sp2)–F bond activation thus succeeded via an unusual Ni0-mediated SNAr. Surprisingly, results show the activation energy (21.8 kcal mol−1) for nucleophilic attack by the Ni0 center is significantly lower than that (36.2 kcal mol−1) by the (SiEt3)− anion. Furthermore, the outer-sphere transition state is more stable than the well-known inner-sphere one because the radius of K+ is too large to form a multicomponent ring with synergistic stabilization. In addition to the Ni⋯Si coordination, this unprecedented transition state in the C(sp2)–F bond activation is stabilized through multiple non-covalent interactions. Additionally, AIMD simulations were employed to elucidate the dynamic effects of the Ni0(silyl)-ate complex initiated C(sp2)–F bond activation, highlighting the pivotal role of multiple non-covalent interactions in the reaction.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
