Asma Marzouk, Konstantinos D. Papavasileiou, Loukas D. Peristeras, Leendert Bezemer, Alexander P. van Bavel, Prathamesh M. Shenai, Ioannis G. Economou
{"title":"A systematic DFT study of structure and electronic properties of titanium dioxide","authors":"Asma Marzouk, Konstantinos D. Papavasileiou, Loukas D. Peristeras, Leendert Bezemer, Alexander P. van Bavel, Prathamesh M. Shenai, Ioannis G. Economou","doi":"10.1002/jcc.27376","DOIUrl":null,"url":null,"abstract":"<p>DFT functionals are of paramount importance for an accurate electronic and structural description of transition metal systems. In this work, a systematic analysis using some well-known and commonly used DFT functionals is performed. A comparison of the structural and energetic parameters calculated with the available experimental data is made in order to find the adequate functional for an accurate description of the TiO<sub>2</sub> bulk and surface of both anatase and rutile structures. In the absence of experimental data on the surface energy, the theoretical predictions obtained using the high-accuracy HSE06 functional were used as a reference to compare against the surface energy values calculated with the other DFT functionals. A clear improvement in the electronic description of both anatase and rutile was observed by introducing the <i>Hubbard U</i> correction term to PBE, PW91, and OptPBE functionals. The OptPBE-<i>U</i>4 functional was found to offer a good compromise between accurately describing the structural and electronic properties of titania.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 25","pages":"2153-2166"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27376","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27376","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
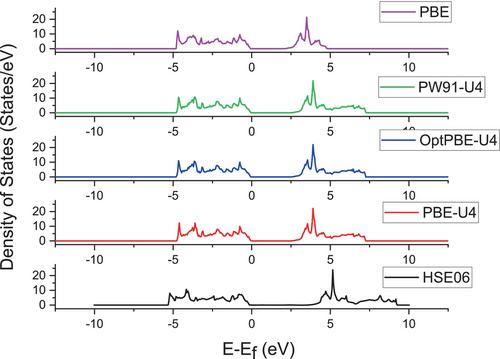
DFT functionals are of paramount importance for an accurate electronic and structural description of transition metal systems. In this work, a systematic analysis using some well-known and commonly used DFT functionals is performed. A comparison of the structural and energetic parameters calculated with the available experimental data is made in order to find the adequate functional for an accurate description of the TiO2 bulk and surface of both anatase and rutile structures. In the absence of experimental data on the surface energy, the theoretical predictions obtained using the high-accuracy HSE06 functional were used as a reference to compare against the surface energy values calculated with the other DFT functionals. A clear improvement in the electronic description of both anatase and rutile was observed by introducing the Hubbard U correction term to PBE, PW91, and OptPBE functionals. The OptPBE-U4 functional was found to offer a good compromise between accurately describing the structural and electronic properties of titania.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
