Edgar Manriquez-Sandoval, Joy Brewer, Gabriela Lule, Samanta Lopez and Stephen D. Fried*,
{"title":"FLiPPR: A Processor for Limited Proteolysis (LiP) Mass Spectrometry Data Sets Built on FragPipe","authors":"Edgar Manriquez-Sandoval, Joy Brewer, Gabriela Lule, Samanta Lopez and Stephen D. Fried*, ","doi":"10.1021/acs.jproteome.3c00887","DOIUrl":null,"url":null,"abstract":"<p >Here, we present FLiPPR, or FragPipe LiP (limited proteolysis) Processor, a tool that facilitates the analysis of data from limited proteolysis mass spectrometry (LiP-MS) experiments following primary search and quantification in FragPipe. LiP-MS has emerged as a method that can provide proteome-wide information on protein structure and has been applied to a range of biological and biophysical questions. Although LiP-MS can be carried out with standard laboratory reagents and mass spectrometers, analyzing the data can be slow and poses unique challenges compared to typical quantitative proteomics workflows. To address this, we leverage FragPipe and then process its output in FLiPPR. FLiPPR formalizes a specific data imputation heuristic that carefully uses missing data in LiP-MS experiments to report on the most significant structural changes. Moreover, FLiPPR introduces a data merging scheme and a protein-centric multiple hypothesis correction scheme, enabling processed LiP-MS data sets to be more robust and less redundant. These improvements strengthen statistical trends when previously published data are reanalyzed with the FragPipe/FLiPPR workflow. We hope that FLiPPR will lower the barrier for more users to adopt LiP-MS, standardize statistical procedures for LiP-MS data analysis, and systematize output to facilitate eventual larger-scale integration of LiP-MS data.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":"23 7","pages":"2332–2342"},"PeriodicalIF":3.6000,"publicationDate":"2024-05-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00887","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
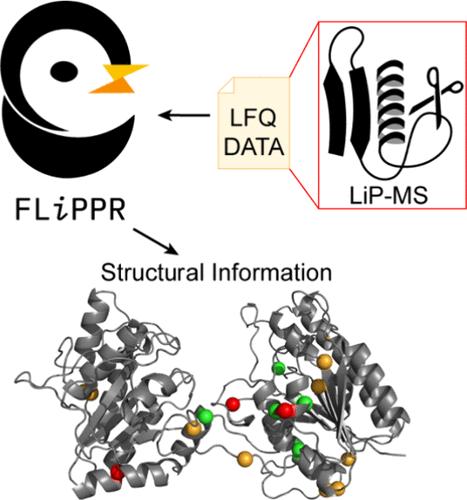
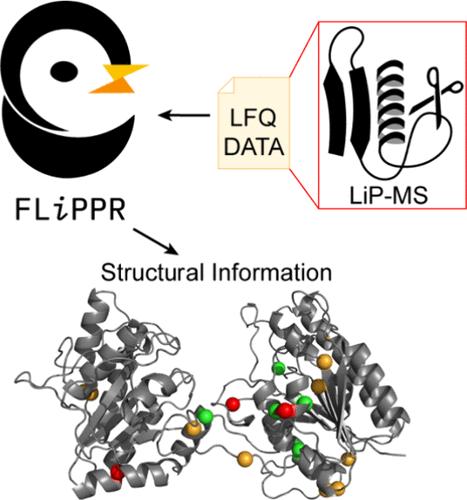
Here, we present FLiPPR, or FragPipe LiP (limited proteolysis) Processor, a tool that facilitates the analysis of data from limited proteolysis mass spectrometry (LiP-MS) experiments following primary search and quantification in FragPipe. LiP-MS has emerged as a method that can provide proteome-wide information on protein structure and has been applied to a range of biological and biophysical questions. Although LiP-MS can be carried out with standard laboratory reagents and mass spectrometers, analyzing the data can be slow and poses unique challenges compared to typical quantitative proteomics workflows. To address this, we leverage FragPipe and then process its output in FLiPPR. FLiPPR formalizes a specific data imputation heuristic that carefully uses missing data in LiP-MS experiments to report on the most significant structural changes. Moreover, FLiPPR introduces a data merging scheme and a protein-centric multiple hypothesis correction scheme, enabling processed LiP-MS data sets to be more robust and less redundant. These improvements strengthen statistical trends when previously published data are reanalyzed with the FragPipe/FLiPPR workflow. We hope that FLiPPR will lower the barrier for more users to adopt LiP-MS, standardize statistical procedures for LiP-MS data analysis, and systematize output to facilitate eventual larger-scale integration of LiP-MS data.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
