Antoine Salichon, Agustin Salcedo, Carine Michel, David Loffreda
{"title":"Theoretical study of structure sensitivity on ceria-supported single platinum atoms and its influence on carbon monoxide adsorption","authors":"Antoine Salichon, Agustin Salcedo, Carine Michel, David Loffreda","doi":"10.1002/jcc.27393","DOIUrl":null,"url":null,"abstract":"<p>Density functional theory (DFT) calculations explore the stability of a single platinum atom on various flat, stepped, and defective ceria surfaces, in the context of single-atom catalysts (SACs) for the water–gas shift (WGS) reaction. The adsorption properties and diffusion kinetics of the metal strongly depend on the support termination with large stability on metastable and stepped CeO<sub>2</sub>(100) and (210) surfaces where the diffusion of the platinum atom is hindered. At the opposite, the more stable CeO<sub>2</sub>(111) and (110) terminations weakly bind the platinum atom and can promote the growth of metallic clusters thanks to fast diffusion kinetics. The adsorption of carbon monoxide on the single platinum atom supported on the various ceria terminations is also sensitive to the surface structure. Carbon monoxide weakly binds to the single platinum atom supported on reduced CeO<sub>2</sub>(111) and (211) terminations. The desorption of the CO<sub>2</sub> formed during the WGS reaction is thus facilitated on the latter terminations. A vibrational analysis underlines the significant changes in the calculated scaled anharmonic CO stretching frequency on these catalysts.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 25","pages":"2167-2179"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27393","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27393","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
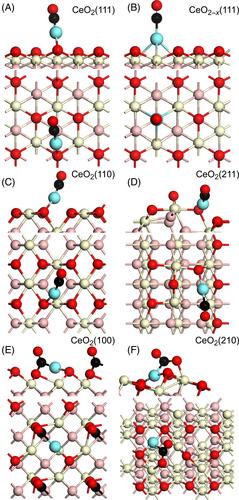
Density functional theory (DFT) calculations explore the stability of a single platinum atom on various flat, stepped, and defective ceria surfaces, in the context of single-atom catalysts (SACs) for the water–gas shift (WGS) reaction. The adsorption properties and diffusion kinetics of the metal strongly depend on the support termination with large stability on metastable and stepped CeO2(100) and (210) surfaces where the diffusion of the platinum atom is hindered. At the opposite, the more stable CeO2(111) and (110) terminations weakly bind the platinum atom and can promote the growth of metallic clusters thanks to fast diffusion kinetics. The adsorption of carbon monoxide on the single platinum atom supported on the various ceria terminations is also sensitive to the surface structure. Carbon monoxide weakly binds to the single platinum atom supported on reduced CeO2(111) and (211) terminations. The desorption of the CO2 formed during the WGS reaction is thus facilitated on the latter terminations. A vibrational analysis underlines the significant changes in the calculated scaled anharmonic CO stretching frequency on these catalysts.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
