{"title":"Effective prediction of SnO2 conduction band edge potential: The key role of surface oxygen vacancies","authors":"Gennaro Vincenzo Sannino, Adriana Pecoraro, Paola Delli Veneri, Michele Pavone, Ana Belén Muñoz-García","doi":"10.1002/jcc.27434","DOIUrl":null,"url":null,"abstract":"<p>Several theoretical studies at different levels of theory have attempted to calculate the absolute position of the SnO<sub>2</sub> conduction band, whose knowledge is key for its effective application in optoelectronic devices such us, for example, perovskite solar cells. However, the predicted band edges fall outside the experimentally measured range. In this work, we introduce a computational scheme designed to calculate the conduction band minimum values of SnO<sub>2</sub>, yielding results aligned with experiments. Our analysis points out the fundamental role of encompassing surface oxygen vacancies to properly describe the electronic profile of this material. We explore the impact of both bridge and in-plane oxygen vacancy defects on the structural and electronic properties of SnO<sub>2</sub>, explaining from an atomistic perspective the experimental observables. The results underscore the importance of simulating both types of defects to accurately predict SnO<sub>2</sub> features and provide new fundamental insights that can guide future studies concerning design and optimization of SnO<sub>2</sub>-based materials and functional interfaces.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 26","pages":"2198-2203"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27434","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
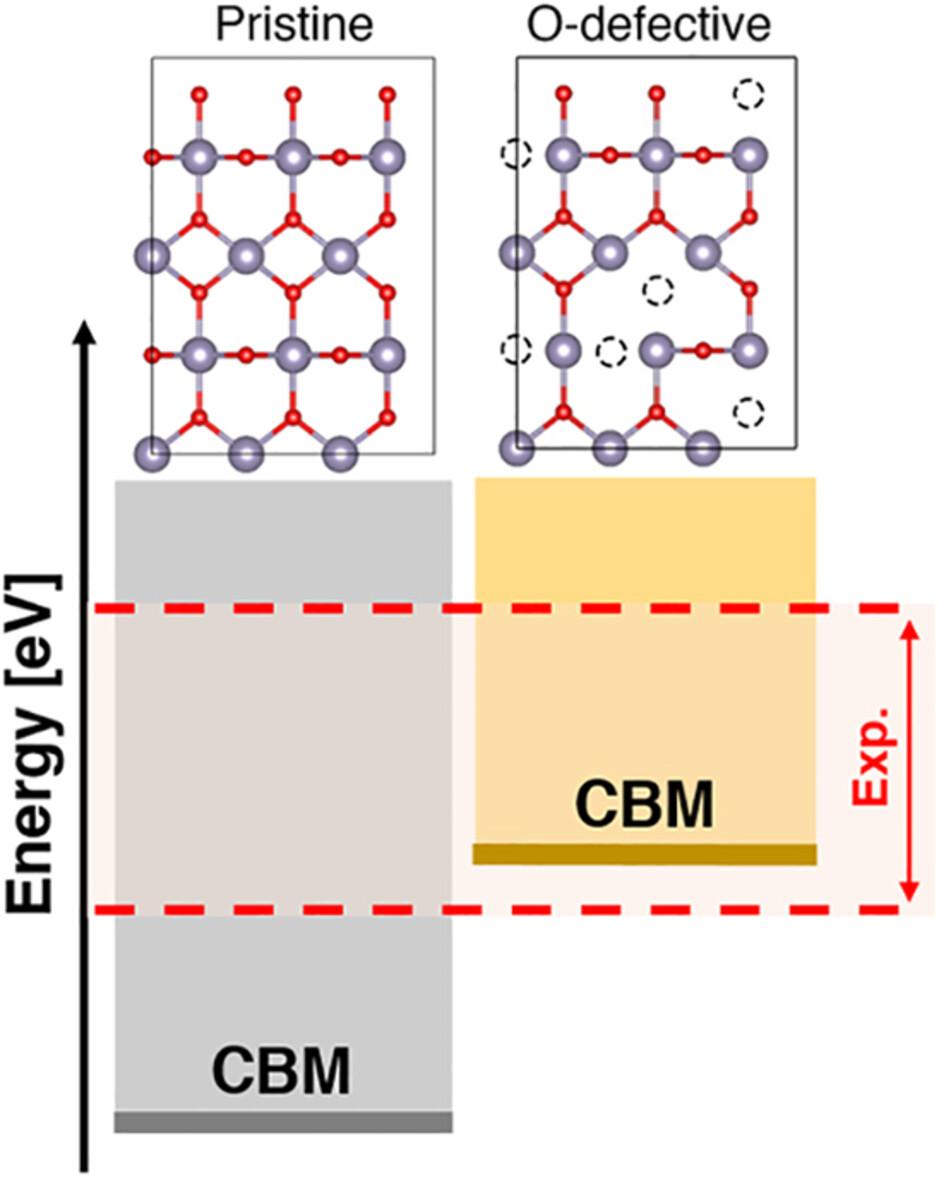
Several theoretical studies at different levels of theory have attempted to calculate the absolute position of the SnO2 conduction band, whose knowledge is key for its effective application in optoelectronic devices such us, for example, perovskite solar cells. However, the predicted band edges fall outside the experimentally measured range. In this work, we introduce a computational scheme designed to calculate the conduction band minimum values of SnO2, yielding results aligned with experiments. Our analysis points out the fundamental role of encompassing surface oxygen vacancies to properly describe the electronic profile of this material. We explore the impact of both bridge and in-plane oxygen vacancy defects on the structural and electronic properties of SnO2, explaining from an atomistic perspective the experimental observables. The results underscore the importance of simulating both types of defects to accurately predict SnO2 features and provide new fundamental insights that can guide future studies concerning design and optimization of SnO2-based materials and functional interfaces.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
