Blake M Hauser, Yuyang Luo, Anusha Nathan, Ahmad Al-Moujahed, Demetrios G Vavvas, Jason Comander, Eric A Pierce, Emily M Place, Kinga M Bujakowska, Gaurav D Gaiha, Elizabeth J Rossin
{"title":"Structure-based network analysis predicts pathogenic variants in human proteins associated with inherited retinal disease.","authors":"Blake M Hauser, Yuyang Luo, Anusha Nathan, Ahmad Al-Moujahed, Demetrios G Vavvas, Jason Comander, Eric A Pierce, Emily M Place, Kinga M Bujakowska, Gaurav D Gaiha, Elizabeth J Rossin","doi":"10.1038/s41525-024-00416-w","DOIUrl":null,"url":null,"abstract":"<p><p>Advances in gene sequencing technologies have accelerated the identification of genetic variants, but better tools are needed to understand which are causal of disease. This would be particularly useful in fields where gene therapy is a potential therapeutic modality for a disease-causing variant such as inherited retinal disease (IRD). Here, we apply structure-based network analysis (SBNA), which has been successfully utilized to identify variant-constrained amino acid residues in viral proteins, to identify residues that may cause IRD if subject to missense mutation. SBNA is based entirely on structural first principles and is not fit to specific outcome data, which makes it distinct from other contemporary missense prediction tools. In 4 well-studied human disease-associated proteins (BRCA1, HRAS, PTEN, and ERK2) with high-quality structural data, we find that SBNA scores correlate strongly with deep mutagenesis data. When applied to 47 IRD genes with available high-quality crystal structure data, SBNA scores reliably identified disease-causing variants according to phenotype definitions from the ClinVar database. Finally, we applied this approach to 63 patients at Massachusetts Eye and Ear (MEE) with IRD but for whom no genetic cause had been identified. Untrained models built using SBNA scores and BLOSUM62 scores for IRD-associated genes successfully predicted the pathogenicity of novel variants (AUC = 0.851), allowing us to identify likely causative disease variants in 40 IRD patients. Model performance was further augmented by incorporating orthogonal data from EVE scores (AUC = 0.927), which are based on evolutionary multiple sequence alignments. In conclusion, SBNA can used to successfully identify variants as causal of disease in human proteins and may help predict variants causative of IRD in an unbiased fashion.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"31"},"PeriodicalIF":4.8000,"publicationDate":"2024-05-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11130145/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-024-00416-w","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
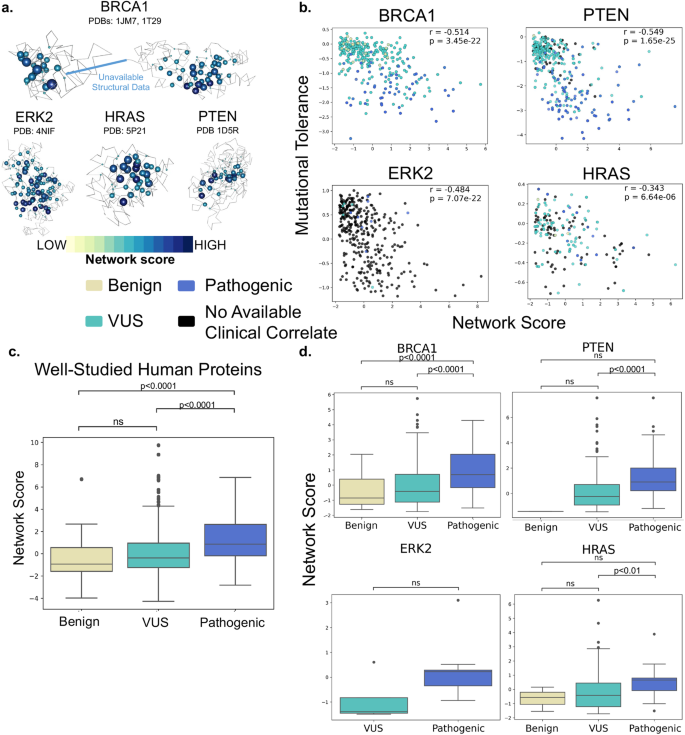
Advances in gene sequencing technologies have accelerated the identification of genetic variants, but better tools are needed to understand which are causal of disease. This would be particularly useful in fields where gene therapy is a potential therapeutic modality for a disease-causing variant such as inherited retinal disease (IRD). Here, we apply structure-based network analysis (SBNA), which has been successfully utilized to identify variant-constrained amino acid residues in viral proteins, to identify residues that may cause IRD if subject to missense mutation. SBNA is based entirely on structural first principles and is not fit to specific outcome data, which makes it distinct from other contemporary missense prediction tools. In 4 well-studied human disease-associated proteins (BRCA1, HRAS, PTEN, and ERK2) with high-quality structural data, we find that SBNA scores correlate strongly with deep mutagenesis data. When applied to 47 IRD genes with available high-quality crystal structure data, SBNA scores reliably identified disease-causing variants according to phenotype definitions from the ClinVar database. Finally, we applied this approach to 63 patients at Massachusetts Eye and Ear (MEE) with IRD but for whom no genetic cause had been identified. Untrained models built using SBNA scores and BLOSUM62 scores for IRD-associated genes successfully predicted the pathogenicity of novel variants (AUC = 0.851), allowing us to identify likely causative disease variants in 40 IRD patients. Model performance was further augmented by incorporating orthogonal data from EVE scores (AUC = 0.927), which are based on evolutionary multiple sequence alignments. In conclusion, SBNA can used to successfully identify variants as causal of disease in human proteins and may help predict variants causative of IRD in an unbiased fashion.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
