Kinan Alhallak, Jun Nagai, Kendall Zaleski, Sofia Marshall, Tamara Salloum, Tahereh Derakhshan, Hiroaki Hayashi, Chunli Feng, Radomir Kratchmarov, Juying Lai, Virinchi Kuchibhotla, Airi Nishida, Barbara Balestrieri, Tanya Laidlaw, Daniel F. Dwyer, Joshua A. Boyce
{"title":"Mast cells control lung type 2 inflammation via prostaglandin E2-driven soluble ST2","authors":"Kinan Alhallak, Jun Nagai, Kendall Zaleski, Sofia Marshall, Tamara Salloum, Tahereh Derakhshan, Hiroaki Hayashi, Chunli Feng, Radomir Kratchmarov, Juying Lai, Virinchi Kuchibhotla, Airi Nishida, Barbara Balestrieri, Tanya Laidlaw, Daniel F. Dwyer, Joshua A. Boyce","doi":"10.1016/j.immuni.2024.05.003","DOIUrl":null,"url":null,"abstract":"<p>Severe asthma and sinus disease are consequences of type 2 inflammation (T2I), mediated by interleukin (IL)-33 signaling through its membrane-bound receptor, ST2. Soluble (s)ST2 reduces available IL-33 and limits T2I, but little is known about its regulation. We demonstrate that prostaglandin E<sub>2</sub> (PGE<sub>2</sub>) drives production of sST2 to limit features of lung T2I. PGE<sub>2</sub>-deficient mice display diminished sST2. In humans with severe respiratory T2I, urinary PGE<sub>2</sub> metabolites correlate with serum sST2. In mice, PGE<sub>2</sub> enhanced sST2 secretion by mast cells (MCs). Mice lacking MCs, ST2 expression by MCs, or E prostanoid (EP)<sub>2</sub> receptors by MCs showed reduced sST2 lung concentrations and strong T2I. Recombinant sST2 reduced T2I in mice lacking PGE<sub>2</sub> or ST2 expression by MCs back to control levels. PGE<sub>2</sub> deficiency also reversed the hyperinflammatory phenotype in mice lacking ST2 expression by MCs. PGE<sub>2</sub> thus suppresses T2I through MC-derived sST2, explaining the severe T2I observed in low PGE<sub>2</sub> states.</p>","PeriodicalId":13269,"journal":{"name":"Immunity","volume":"22 1","pages":""},"PeriodicalIF":26.3000,"publicationDate":"2024-05-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Immunity","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1016/j.immuni.2024.05.003","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
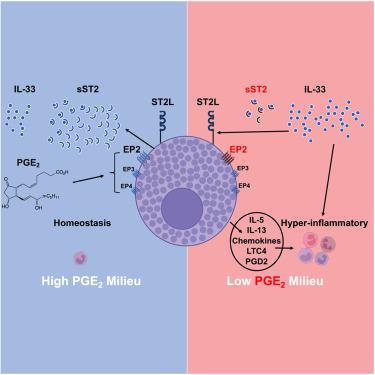
Severe asthma and sinus disease are consequences of type 2 inflammation (T2I), mediated by interleukin (IL)-33 signaling through its membrane-bound receptor, ST2. Soluble (s)ST2 reduces available IL-33 and limits T2I, but little is known about its regulation. We demonstrate that prostaglandin E2 (PGE2) drives production of sST2 to limit features of lung T2I. PGE2-deficient mice display diminished sST2. In humans with severe respiratory T2I, urinary PGE2 metabolites correlate with serum sST2. In mice, PGE2 enhanced sST2 secretion by mast cells (MCs). Mice lacking MCs, ST2 expression by MCs, or E prostanoid (EP)2 receptors by MCs showed reduced sST2 lung concentrations and strong T2I. Recombinant sST2 reduced T2I in mice lacking PGE2 or ST2 expression by MCs back to control levels. PGE2 deficiency also reversed the hyperinflammatory phenotype in mice lacking ST2 expression by MCs. PGE2 thus suppresses T2I through MC-derived sST2, explaining the severe T2I observed in low PGE2 states.
期刊介绍:
Immunity is a publication that focuses on publishing significant advancements in research related to immunology. We encourage the submission of studies that offer groundbreaking immunological discoveries, whether at the molecular, cellular, or whole organism level. Topics of interest encompass a wide range, such as cancer, infectious diseases, neuroimmunology, autoimmune diseases, allergies, mucosal immunity, metabolic diseases, and homeostasis.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
