Christoph Bueschl, Gabriel Riquelme, Nicolás Zabalegui, Maximilian A Rey, María Eugenia Monge
{"title":"Tidy-Direct-to-MS: An Open-Source Data-Processing Pipeline for Direct Mass Spectrometry-Based Metabolomics Experiments.","authors":"Christoph Bueschl, Gabriel Riquelme, Nicolás Zabalegui, Maximilian A Rey, María Eugenia Monge","doi":"10.1021/acs.jproteome.3c00784","DOIUrl":null,"url":null,"abstract":"<p><p>Direct-to-Mass Spectrometry and ambient ionization techniques can be used for biochemical fingerprinting in a fast way. Data processing is typically accomplished with vendor-provided software tools. Here, a novel, open-source functionality, entitled Tidy-Direct-to-MS, was developed for data processing of direct-to-MS data sets. It allows for fast and user-friendly processing using different modules for optional sample position detection and separation, mass-to-charge ratio drift detection and correction, consensus spectra calculation, and bracketing across sample positions as well as feature abundance calculation. The tool also provides functionality for the automated comparison of different sets of parameters, thereby assisting the user in the complex task of finding an optimal combination to maximize the total number of detected features while also checking for the detection of user-provided reference features. In addition, Tidy-Direct-to-MS has the capability for data quality review and subsequent data analysis, thereby simplifying the workflow of untargeted ambient MS-based metabolomics studies. Tidy-Direct-to-MS is implemented in the Python programming language as part of the TidyMS library and can thus be easily extended. Capabilities of Tidy-Direct-to-MS are showcased in a data set acquired in a marine metabolomics study reported in MetaboLights (MTBLS1198) using a transmission mode Direct Analysis in Real Time-Mass Spectrometry (TM-DART-MS)-based method.</p>","PeriodicalId":48,"journal":{"name":"Journal of Proteome Research","volume":null,"pages":null},"PeriodicalIF":3.8000,"publicationDate":"2024-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Proteome Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1021/acs.jproteome.3c00784","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
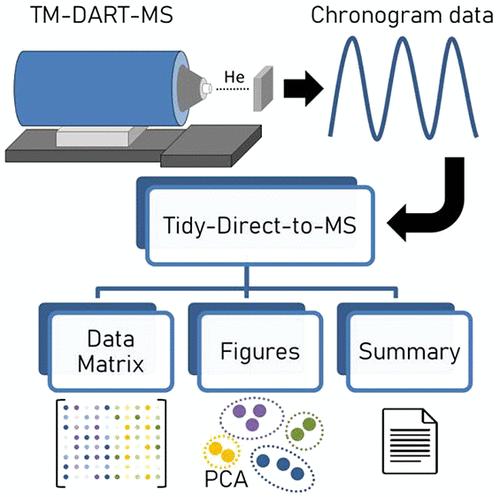
Direct-to-Mass Spectrometry and ambient ionization techniques can be used for biochemical fingerprinting in a fast way. Data processing is typically accomplished with vendor-provided software tools. Here, a novel, open-source functionality, entitled Tidy-Direct-to-MS, was developed for data processing of direct-to-MS data sets. It allows for fast and user-friendly processing using different modules for optional sample position detection and separation, mass-to-charge ratio drift detection and correction, consensus spectra calculation, and bracketing across sample positions as well as feature abundance calculation. The tool also provides functionality for the automated comparison of different sets of parameters, thereby assisting the user in the complex task of finding an optimal combination to maximize the total number of detected features while also checking for the detection of user-provided reference features. In addition, Tidy-Direct-to-MS has the capability for data quality review and subsequent data analysis, thereby simplifying the workflow of untargeted ambient MS-based metabolomics studies. Tidy-Direct-to-MS is implemented in the Python programming language as part of the TidyMS library and can thus be easily extended. Capabilities of Tidy-Direct-to-MS are showcased in a data set acquired in a marine metabolomics study reported in MetaboLights (MTBLS1198) using a transmission mode Direct Analysis in Real Time-Mass Spectrometry (TM-DART-MS)-based method.
期刊介绍:
Journal of Proteome Research publishes content encompassing all aspects of global protein analysis and function, including the dynamic aspects of genomics, spatio-temporal proteomics, metabonomics and metabolomics, clinical and agricultural proteomics, as well as advances in methodology including bioinformatics. The theme and emphasis is on a multidisciplinary approach to the life sciences through the synergy between the different types of "omics".



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
