Jie Li, Oufan Zhang, Kunyang Sun, Yingze Wang, Xingyi Guan, Dorian Bagni, Mojtaba Haghighatlari, Fiona L Kearns, Conor Parks, Rommie E Amaro, Teresa Head-Gordon
{"title":"Mining for Potent Inhibitors through Artificial Intelligence and Physics: A Unified Methodology for Ligand Based and Structure Based Drug Design.","authors":"Jie Li, Oufan Zhang, Kunyang Sun, Yingze Wang, Xingyi Guan, Dorian Bagni, Mojtaba Haghighatlari, Fiona L Kearns, Conor Parks, Rommie E Amaro, Teresa Head-Gordon","doi":"10.1021/acs.jcim.4c00634","DOIUrl":null,"url":null,"abstract":"<p><p>Determining the viability of a new drug molecule is a time- and resource-intensive task that makes computer-aided assessments a vital approach to rapid drug discovery. Here we develop a machine learning algorithm, iMiner, that generates novel inhibitor molecules for target proteins by combining deep reinforcement learning with real-time 3D molecular docking using AutoDock Vina, thereby simultaneously creating chemical novelty while constraining molecules for shape and molecular compatibility with target active sites. Moreover, through the use of various types of reward functions, we have introduced novelty in generative tasks for new molecules such as chemical similarity to a target ligand, molecules grown from known protein bound fragments, and creation of molecules that enforce interactions with target residues in the protein active site. The iMiner algorithm is embedded in a composite workflow that filters out Pan-assay interference compounds, Lipinski rule violations, uncommon structures in medicinal chemistry, and poor synthetic accessibility with options for cross-validation against other docking scoring functions and automation of a molecular dynamics simulation to measure pose stability. We also allow users to define a set of rules for the structures they would like to exclude during the training process and postfiltering steps. Because our approach relies only on the structure of the target protein, iMiner can be easily adapted for the future development of other inhibitors or small molecule therapeutics of any target protein.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"9082-9097"},"PeriodicalIF":5.3000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11683870/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00634","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/6 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
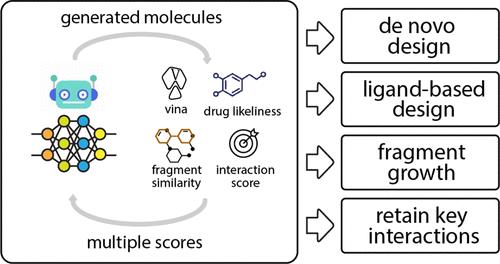
Determining the viability of a new drug molecule is a time- and resource-intensive task that makes computer-aided assessments a vital approach to rapid drug discovery. Here we develop a machine learning algorithm, iMiner, that generates novel inhibitor molecules for target proteins by combining deep reinforcement learning with real-time 3D molecular docking using AutoDock Vina, thereby simultaneously creating chemical novelty while constraining molecules for shape and molecular compatibility with target active sites. Moreover, through the use of various types of reward functions, we have introduced novelty in generative tasks for new molecules such as chemical similarity to a target ligand, molecules grown from known protein bound fragments, and creation of molecules that enforce interactions with target residues in the protein active site. The iMiner algorithm is embedded in a composite workflow that filters out Pan-assay interference compounds, Lipinski rule violations, uncommon structures in medicinal chemistry, and poor synthetic accessibility with options for cross-validation against other docking scoring functions and automation of a molecular dynamics simulation to measure pose stability. We also allow users to define a set of rules for the structures they would like to exclude during the training process and postfiltering steps. Because our approach relies only on the structure of the target protein, iMiner can be easily adapted for the future development of other inhibitors or small molecule therapeutics of any target protein.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
