Shaoze Zhang, De-en Jiang, Nan Zhou, Jiaxing Tang, Keyu Zhang, Yin Li, Junxian Hu, Changjun Peng, Honglai Liu, Bin Yang, Yaochun Yao
{"title":"Ionic liquids intercalation in titanium carbide MXenes: A first-principles investigation","authors":"Shaoze Zhang, De-en Jiang, Nan Zhou, Jiaxing Tang, Keyu Zhang, Yin Li, Junxian Hu, Changjun Peng, Honglai Liu, Bin Yang, Yaochun Yao","doi":"10.1002/jcc.27444","DOIUrl":null,"url":null,"abstract":"<p>Herein, we present a density functional theory with dispersion correction (DFT-D) calculations that focus on the intercalation of ionic liquids (ILs) electrolytes into the two-dimensional (2D) Ti<sub>3</sub>C<sub>2</sub>T<sub>x</sub> MXenes. These ILs include the cation 1-ethyl-3-methylimidazolium (Emim<sup>+</sup>), accompanied by three distinct anions: bis(trifluoromethylsulfonyl)imide (TFSA<sup>−</sup>), (fluorosulfonyl)imide (FSA<sup>−</sup>) and fluorosulfonyl(trifluoromethanesulfonyl)imide (FTFSA<sup>−</sup>). By altering the surface termination elements, we explore the intricate geometries of IL intercalation in neutral, negative, and positive pore systems. Accurate estimation of charge transfer is achieved through five population analysis models, such as Hirshfeld, Hirshfeld-I, DDEC6 (density derived electrostatic and chemical), Bader, and VDD (voronoi deformation density) charges. In this work, we recommend the DDEC6 and Hirshfeld-I charge models, as they offer moderate values and exhibit reasonable trends. The investigation, aimed at visualizing non-covalent interactions, elucidates the role of cation-MXene and anion-MXene interactions in governing the intercalation phenomenon of ionic liquids within MXenes. The magnitude of this role depends on two factors: the specific arrangement of the cation, and the nature of the anionic species involved in the process.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 27","pages":"2294-2307"},"PeriodicalIF":4.8000,"publicationDate":"2024-06-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27444","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
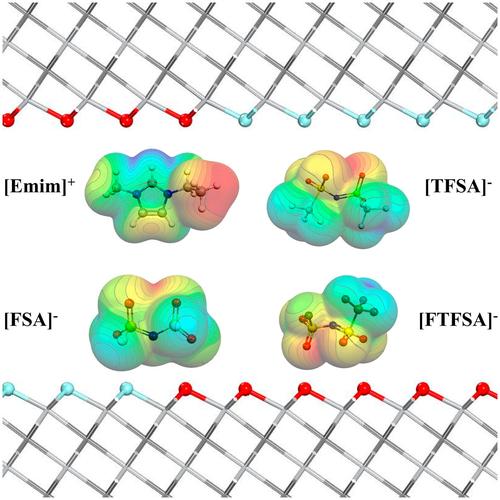
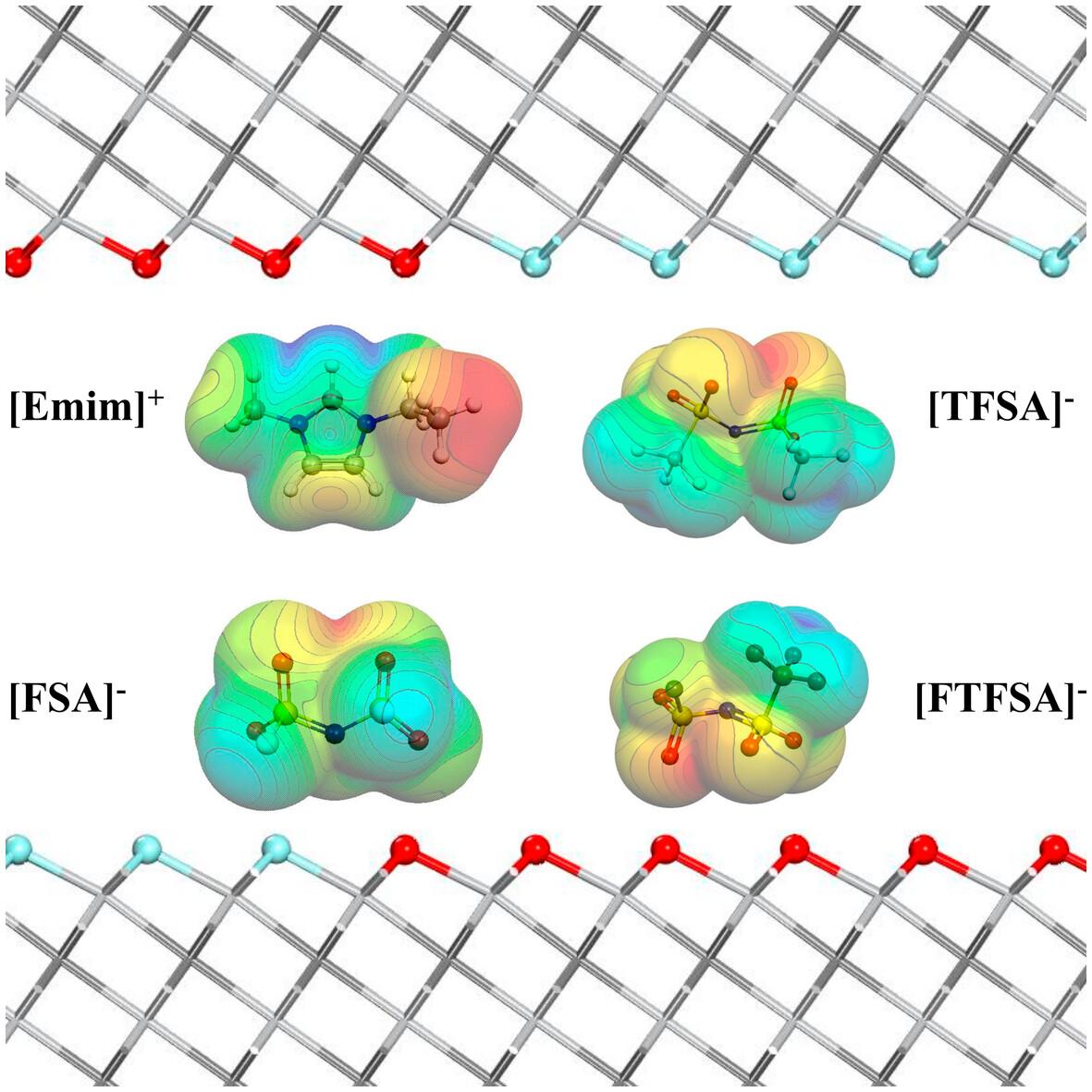
Herein, we present a density functional theory with dispersion correction (DFT-D) calculations that focus on the intercalation of ionic liquids (ILs) electrolytes into the two-dimensional (2D) Ti3C2Tx MXenes. These ILs include the cation 1-ethyl-3-methylimidazolium (Emim+), accompanied by three distinct anions: bis(trifluoromethylsulfonyl)imide (TFSA−), (fluorosulfonyl)imide (FSA−) and fluorosulfonyl(trifluoromethanesulfonyl)imide (FTFSA−). By altering the surface termination elements, we explore the intricate geometries of IL intercalation in neutral, negative, and positive pore systems. Accurate estimation of charge transfer is achieved through five population analysis models, such as Hirshfeld, Hirshfeld-I, DDEC6 (density derived electrostatic and chemical), Bader, and VDD (voronoi deformation density) charges. In this work, we recommend the DDEC6 and Hirshfeld-I charge models, as they offer moderate values and exhibit reasonable trends. The investigation, aimed at visualizing non-covalent interactions, elucidates the role of cation-MXene and anion-MXene interactions in governing the intercalation phenomenon of ionic liquids within MXenes. The magnitude of this role depends on two factors: the specific arrangement of the cation, and the nature of the anionic species involved in the process.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
